- Lay summary
- Executive summary
- 1. Introduction
- 2. Literature Search
- 3. Expert stakeholder perspectives on the adoption of NAMs into regulations
- 3.1. Stakeholder Interviews: Methodology
- 3.2 Key findings from the stakeholder interviews
- 3.2.1. Opinions on the use of the acronym NAM
- 3.2.2. Views on the scale and direction of investment into NAM research
- 3.2.3. Perspectives on which NAMs are the focus of investment into research
- 3.2.4. Current approaches towards the integration of NAMs into regulatory use
- 3.2.5. Comments on how NAM integration will be done in the future
- 3.2.6. Regulatory activities where NAMs can play a key role into the future
- 3.2.7. Barriers to NAM integration into regulations
- 3.2.8. Emerging substances and materials where NAMs are well suited to play a part in their risk assessment
- 4. Summary of literature search: Wider perspectives on NAMs and their integration into regulations
- 4.1. Criticisms of the existing regulatory toxicology paradigm
- 4.2. If not animal studies, then what?
- 4.3. Considerations on the strengths and weaknesses of the AOP approach
- 4.4. Use of IATA and Defined Approaches (DA) in regulatory assessments
- 4.5. Context of Use
- 4.6. Use of NAMs to identify low toxicity substances
- 4.7. National and regional strategies to further integrate NAMs into regulations
- 5. Perspectives on specific NAMs
- 5.1. Regulatory Readiness Assessment of NAMs: a general discussion
- 5.2. In Silico methods
- 5.2.1. Considerations for Regulatory Readiness Specific to In Silico Models
- 5.2.2. Expert opinions of the Regulatory Readiness of In Silico Models
- QSAR models
- 5.3. Regulatory Readiness of In Vitro and In Chemico Test Methods
- 5.3.1. Considerations for Regulatory Readiness Specific to In Vitro and In Chemico Test Methods
- 5.3.2. Expert views on the regulatory readiness of in vitro and in chemico test methods
- 5.4. Regulatory Readiness of omics technologies
- 5.5. Regulatory Readiness of Grouping and Read-Across (RxA)
- 5.6. Regulatory Readiness of Other NAMs
- 6. The use of NAMs in regulation
- 7. Perspectives on the use of NAMs in risk assessments related to food
- 8. Conclusion and next steps
- Annexes
Lay summary
The use of animal studies to predict the health impacts for humans from exposure to chemicals and to guide the regulations that attempt to minimise these impacts have been an undesirable but necessary activity across the globe. This has been because non-animal alternatives are generally regarded as not providing sufficient information. The development of these New Approach Methodologies (NAMs) is the goal of a huge area of research and they are starting to play an increasing role in regulations, but usually to predict very specific health issues. The UK Food Standards Agency (FSA) has recently published a roadmap describing their plans to use NAMs to ensure the safety of food in the UK. The UK is at the forefront of research into NAMs with globally renowned experts in academia, regulatory bodies, private companies and contract research organisations. This means it is well-positioned to play a leading role in progress towards the reduction, refinement and replacement of animal studies in regulations internationally.
This report gives a summary of how NAMs are being used in regulations, the efforts being made by different regions to further integrate NAMs into regulations. It also presents the opinions of experts on the topic about the speed and direction of this integration both through an extensive literature search and interviews with 20 stakeholders from different global regions. The term NAM covers a wide variety of technologies and this report describes how close decision-makers believe each technology is to playing a part in regulatory chemical risk assessment. It describes the reasons why regulators may not have confidence in using various NAMs in their decision-making process and the improvements that can be made to increase confidence. The UK FSA roadmap aligns with other initiatives from around the globe, allowing the FSA to use global best practices to refine their approach to risk assessment in the future.
Executive summary
New Approach Methodologies or NAMs is a term introduced in 2016 to cover all technologies that attempt to reduce, refine or replace (the 3Rs) animal studies. The integration of NAMs into chemical regulations is an expressed goal in most regions globally but some stakeholders feel this has been frustratingly slow. The term NAM covers a wide range of technologies and strategies, including in vitro and in chemico studies, in silico modelling, omics, organ-on-a-chip and read-across (RxA) strategies. Whilst some of these are already being used in regulatory processes, they are often to support the interpretation of animal study results or for screening and prioritisation processes rather than as a replacement for animal studies. The UK FSA recently published a roadmap to outline their approach to integrate NAMs into the assessments needed to maintain food safety in the UK. This report presents the results of a literature search on the current use of NAMs in regulations around the world. It also examines the leading roadmaps from the past decade and initiatives by various regulators to further integrate NAMs into their activities. It also outlines the barriers that need to be overcome to accelerate this integration and the suggested opportunities to achieve this through cutting edge research. The opinions taken from the literature search are supplemented by over 20 interviews with expert stakeholders from around the globe.
One of the biggest barriers to the greater use of NAMs is a lack of confidence from regulators in how well NAMs can predict adverse effects when compared to animal studies. It is recognised that a single NAM will not be a direct 1-2-1 replacement of an animal study, with a battery of NAMs being required. While there are still gaps in our understanding of the uncertainties and biological significance of individual NAMs, another challenge is how to define and combine the required set of NAMs to reliably predict their effects on human health. Although there remains gaps in understanding the uncertainties and biological relevance of individual NAMs there is the additional issue around how to define and integrate the necessary group of NAMs to give a reliable prediction of effects to human health. Validation is the accepted way to gain confidence in the reliability of study outcomes with clearly defined biological significance. However, this process can be very time-consuming and for some NAMs may not always be deemed suitable in its current format. Alternative validation approaches have been suggested but their acceptance from regulatory bodies is slow.
In the absence of direct replacement techniques, the use of grouping and read-across (RxA) to use existing animal data to fill data gaps, rather than commissioning new studies, is proposed as one of the most attractive ways to reduce animal testing in the short-term. This approach is accepted in many global regulations although the quality of their justification has been inconsistent. The approach needs a comprehensive scientific justification for which NAMs are well suited. NAMs can identify and justify common Adverse Outcome Pathways (AOPs) between group members and hence support RxA to animal data from data-rich group members. Physiologically Based Kinetic (PBK) models have been identified as a vital part of the AOP approach as they are able to link Points of Departure (PoD) observed at a cellular or organ level in NAMs to dosimetry of the entire organism. Consequently, they have the potential to play a key role in determining safe exposure levels for humans, upon which many existing regulations rely. Some stakeholders believe that this approach could lead to a paradigm shift in regulations that are based directly on the risk to humans rather than the current approach of hazard to animals, a Next Generation Risk Assessment (NGRA), but how quickly this could happen is debatable.
NAMs have already replaced animal studies for some endpoints that address local exposure of a specific organ (e.g. skin sensitisation, eye irritation), but this is not foreseen in the near to medium term for more complex endpoints that address systemic effects to multiple organs. This is in part due to the fact that a network of AOPs is applicable to these endpoints. It is hoped that emerging technologies such as omics and organ-on-a-chip will address this complexity, but this will need the greater use of digital technologies to collect, synthesise and interpret the quantity of data that these studies can generate. Stakeholders recognise that this data will need to be re-usable so researchers will need to follow common reporting processes, such as reporting templates, standards and agreed ontologies, to properly utilise the potential of these methods. International bodies such as the Organisation for Economic Co-operation and Development (OECD) are very active in providing tools and guidance to achieve this goal.
1. Introduction
The field of chemical safety is constantly evolving, and scientific advancements have led to the development, implementation, and acceptance of reliable and relevant new approach methodologies (NAMs). NAMs offer alternatives to traditional animal testing methods for chemical safety assessment and aim to replace, refine, or reduce reliance on such methods. NAMs encompass a broad range of techniques, technologies, and approaches that embrace ethical research principles and are increasingly being used for regulatory decision-making by agencies worldwide. The UK Food Standards Agency (UK FSA) aims to protect public dietary health and consumers’ wider interests in food. UK FSA’s fundamental mission is ‘food you can trust’. This means people can have confidence that the food they purchase and consume is safe, accurately labelled, healthier, and more sustainable. The five year strategy for the UK FSA, published in 2022 (FSA, 2022), is based around three pillars.
-
Food is safe.
-
Food is what it says it is.
-
Food is healthier and more sustainable
At the heart of this mission to ensure food you can trust, is the guiding principle of being driven by science and evidence. This means that all UK FSA’s advice and decisions, including food policy and product authorisations, are based on the latest cutting-edge scientific evidence, which is openly published on the FSA’s website. New technologies, evolving business models, and changing consumer behaviours require the UK FSA to rethink how they deliver this mission. It is felt that NAMs can provide a useful tool to help achieve this goal. In a 2023 roadmap entitled “Paving the way for a UK Roadmap: Development, Endorsement and Regulatory Acceptance of New Approach Methodologies (NAMs) in Chemical Risk Assessment and Beyond”, it was recognised that the journey towards greater inclusion of NAMs into chemical risk assessment needed to “support and initiate research to ensure that the most promising technologies are identified, developed, validated, and integrated.” and to “assess the list of NAMs and other NAMs roadmaps” (UK FSA, 2023). This report is intended to assist in meeting these targets. The objectives of the project were as follows:
-
Conduct a comprehensive literature review to explore recent studies, emerging trends and best use of NAMs in the field of chemical safety
-
Assess the regulatory readiness of the NAMs identified in the comprehensive literature review
-
Understand the usefulness and confidence of NAMs in the regulatory decision-making process
2. Literature Search
2.1. Methodology
The primary goal of this project was to conduct a thorough review of current scientific literature to gather the latest insights into the use of NAMs in chemical risk assessment, with a particular focus on their role in ensuring food safety. Employing a predefined structured approach, we systematically conducted searches, screened materials, made selections, categorised findings, and organised data. Our methodology for the literature review is based on the approach outlined by Collins et al. (2015) to quick scoping reviews and rapid evidence assessment (Collins et al., 2015) . Below, we outline the general process used for identifying relevant literature.
2.1.1. Literature-identification process
STEP 1: Establishing search terms and search strings
In collaboration with the UK FSA, we initiated the process of identifying pertinent keywords aligned with the project’s objectives. This involved identifying relevant search terms, including synonyms and alternate terms, followed by examining the search results in both PubMed and ScienceDirect databases. Terms generating over 3,000 hits were either refined or omitted due to their broad scope (e.g. “artificial intelligence”, “machine learning”, “standardisation”), while those yielding fewer than 3,000 hits were retained for their perceived specificity and direct relevance to the project.
After this scoping exercise, seven categories of search terms were developed:
-
Category A: Set of terms related to generalised NAMs terminology
-
Category B: Set of terms related to NAMs in an Integrated Approaches to Testing and Assessment (IATA) for Next Generation Risk Assessment (NGRA)
-
Category C: Set of terms related to the type of test system
-
Category D: Set of terms related to regulations / compliance
-
Category E: Set of terms related to chemical / product families
-
Category F: Set of terms related to oral exposure
-
Category F: Set of terms related to Food / Food compounds
Categories with over 15 search terms were further divided into subcategories based on terms representing similar concepts or synonyms (see Annex 1) for further details).
Within each category, the Boolean Operator ‘OR’ was used to connect search terms to retrieve results that include at least one of the keywords . Additionally, some search terms consisted of groups of words that were searched as an exact phrase by enclosing the words in double quotation marks (“Next Generation Risk Assessment”) or using a hyphen (“organ-on-a-chips”). Also, in some cases, an asterisk was used as a wildcard character to truncate the phrase and retrieve the root word with alternate endings (“non-animal method*” capturing for example both “methods” and “methodology”).
The following filters were applied for the literature search:
-
Publications from 2014 onwards to prioritise the most recent literature and ensure the relevance of the studies. NAMs published over a decade ago were excluded from the literature review as they are considered either well-established within the regulatory framework or had been superseded by improved methods meaning research into them had halted.
-
Publications in English
After the initial screening process, we conducted multiple literature searches in PubMed, focusing on title and abstract. The goal was to identify the most effective combination of categories, also referred to as ‘search strings,’ which yielded the most relevant references. Initially, we constructed search strings comprising two categories, utilizing the Boolean Operators ‘AND’ to ensure results included search terms from each category in the title or abstract of the paper. The number of hits retrieved from PubMed was noted for each combination. However, as the initial two-category combinations resulted in a substantial number of hits (exceeded 10,000 hits for certain combinations), we proceeded to develop search strings incorporating combinations of three and four categories.
Details regarding the search strings employed to conduct the search strategy for PubMed and the respective description of searched fields are provided in Table 1.
In cases where the number of hits exceeded 300, we employed the Boolean operator ‘NOT’ to exclude terms deemed off-topic, such as “environment,” “ecotox*,” and “drug,” aiming to refine the search results to maintain relevance.
Subsequently, the retrieved search results from PubMed were directly exported into the reference management software EndNote. This process involved creating an EndNote master library containing the exported search string results. Following importation, diligent efforts were made to eliminate any duplicate entries from the master library to ensure data integrity and streamline subsequent analyses.
STEP 2: Literature Screening
Throughout the screening process, a decision had to be made about what types of references would be prioritised as most beneficial to the project’s goals. The screening process was divided into two phases.
During phase 1, title and abstract screening of literature for relevancy was conducted manually by experienced toxicologists. Specific criteria were used to prioritise which references were relevant to this project and which to exclude.
Examples of inclusion criteria were:
-
Literature addressing the application of NAMs in assessing human health aspect
-
NAMs related to various aspects of chemical safety, such as toxicity testing, risk assessment, and hazard identification and, where applicable, more specifically in food safety
-
Studies proposed to be or actively being considered for a standardised protocol by one of the standardisation organisations (for NAMs); or directly aligned with an accepted regulatory approach (for methods that use existing data)
-
Publications specifically addressing regulatory decision processes
-
Research articles, reviews, and reports published in peer-reviewed journals or from regulatory bodies and organisations that play an important role in implementing NAMs and recommended for use in regulatory guidance (for (Quantitative) Structure Activity Relationship ((Q)SAR) models) or in which a relation to a regulatory apical endpoint is clearly explained (for in vitro studies)
Examples of exclusion criteria were:
-
Studies not directly related to NAMs in the context of human health and chemical safety
-
Publications with a primary focus on traditional animal methods without significant incorporation of NAMs
-
Irrelevant articles that don’t contribute to the current understanding of NAMs in chemical safety or regulatory decision processes in the human health context
-
NAMs well established in the regulatory toxicology and regulatory framework (e.g. Ames test and micronucleus test for mutagenicity)
-
NAMs where the applicability domain is not clearly defined (for (Quantitative) structure activity relationships ((Q)SARs))
-
Non-peer-reviewed sources and grey literature lacking detailed information
-
Literature focused on biomedical research, vaccines, drugs, cigarettes, basic research, environmental risk assessment, ecotoxicity, cancer diagnosis, foodborne pathogens detection/quantification, medical devices
During phase 2, full-text screening was conducted to categorise and extract relevant literature. In cases where the primary investigators could not reach a consensus regarding the inclusion of an article, an independent investigator, not directly involved in the initial searches, was consulted to make the final decision. For this step, a specific approach was adopted to determine inclusions.
Initially, a search was performed using the term “regulat*” to analyse how authors aligned their papers with regulatory purposes. It was observed that many authors briefly mentioned regulatory implications in the introduction without further elaboration elsewhere in the paper. Such papers were subsequently excluded from the library. Conversely, papers that explicitly contextualised regulatory frameworks within the discussion, illustrating how their work could fulfil regulatory requirements, were included, even if the methodology hadn’t been fully validated. Numerous papers were identified discussing the application of NAMs in pharmaceutical development even though this was not apparent during the initial screening process. While these might offer insights applicable to chemical risk assessment, it was decided that they fell outside the project’s scope. Moreover, many of these papers primarily focused on assessing the efficacy of a substance as an active pharmaceutical ingredient rather than the toxicity of the substance.
STEP 3: Literature Categorisation and Extraction
Following the completion of the screening phases, toxicologists reviewed full-text literature information, extracting and organising references into an Excel spreadsheet.
To ensure the user-friendliness of the literature library, each identified hit (including source, year, authors, title, publication, DOI) underwent classification based on key aspects of the paper or report. Categories were determined by subdividing overall topics identified during the initial screening process. There were no limitations on the number of categories a paper could be assigned to, as the content of the library ranged from general discussions on the uses of any NAMs to detailed descriptions of an individual NAM for specific substances and endpoints. The goal of this categorisation was to facilitate rapid filtering of relevant papers for users based on their specific needs.
The following categories and subdivisions were developed to categorise the extracted references:
-
Assessment context
-
Hazard
-
Exposure (this is in the context of translating NAM dosing to a specific organ to in vivo dosimetry and covered articles discussing topics such as toxicokinetics)
-
Risk
-
-
Focus of literature
-
Describing an individual NAM
-
Describing a battery of NAMS in an IATA for NGRA
-
Describing an AOP
-
Discussion on NAMs in regulation
-
Guidance document
-
Case studies
-
-
Review literature or workshop review
-
Chemical application (where a paper examines an individual substance, class of substance (e.g. bisphenols) or defined use of a substance (e.g. cosmetics, food, nanomaterials).
-
Type of NAM
-
In vitro
-
In silico (e.g. (Q)SAR, predictive model)
-
Omics
-
Grouping / read-across / Weight-of-evidence (WoE)
-
Database
-
Toxicokinetics (Absorption, Distribution, Metabolism and Excretion (ADME)) / In Vitro to In Vivo Extrapolation (IVIVE) / PBK
-
Other (e.g. organ on a chip; artificial intelligence)
-
-
Apical endpoint addressed (including organ)
-
Aspect of risk assessment examined
-
Mechanism (e.g. Adverse Outcome Pathway (AOP), Key Event (KE), Molecular Initiating Event (MIE)
-
Effect Threshold calculation (e.g. Threshold of Toxicological Concern (TTC), No Observed Adverse Effect Level (NOAEL), Derived No Effect Level (DNEL) etc)
-
Screening
-
NGRA (e.g. examining concepts not used in current toxicology assessments)
-
Systems approach
-
-
Related aspect addressed
-
Validation
-
Data curation and access (e.g. databases and how they are maintained)
-
Communication (e.g. templates used to report data)
-
-
Location of authors
-
US
-
Europe
-
Other
-
The Excel spreadsheet containing the categorised literature findings has been provided alongside this report.
2.1.2. Literature sources
Database search
The search strategy described above was implemented in PubMed due to its capacity to accommodate a larger number of keywords per search and its unique advantage of being readily updated with literature presented online in pre-print versions by various journals, a feature not replicated by Scopus or Web of Science (Falagas et al., 2008). However, PubMed lacks citation analysis capabilities. To complement the literature captured by PubMed, an additional tailored search was conducted on Web of Science and Science Direct to ensure comprehensive coverage.
Approximately 20 reviews were selected (see Annex 2) from the reference library for their relevance in discussing NAMs application in risk assessment and exploring avenues for future advancements. These reviews were individually entered into Web of Science, where a “reference” link provided access to all articles cited within the selected review, alongside another link revealing all articles referencing it. Additionally, the “Related Records” feature facilitated the identification of articles sharing citations with the selected review. Science Direct was utilised for its PlumX Metrics functionality, which incorporates policy citations. All policy citations are presented within a designated tab alongside their respective sources. Users have the capability to navigate to both the policy document citing the research and the originating policy organisation. This search approach aimed to uncover additional literature based on authorship and citation patterns, including publications of significant influence. Consequently, about 90 additional articles were retrieved through this expanded search process.
Grey literature search
One of the key goals of the project is to identify where NAMs have been used to reach regulatory decisions. As these decisions are not always published in academic literature, a list of organisations closely related to making regulatory decisions or to developing NAMs for regulatory purposes was identified and their websites were searched manually. This involved a focus on any pages that were entitled “publications” or “deliverables”. Although the focus was placed on finding published reports, other methods of communication including presentations, conference proceedings and webinars were also identified. The organisations examined are shown in Annex 3.
In summary, due to the complexity, broad coverage and rapid expansion of the NAMs field, there’s a possibility that pertinent and accessible information may have been overlooked during the creation of this literature search and report.
2.2. Literature search results
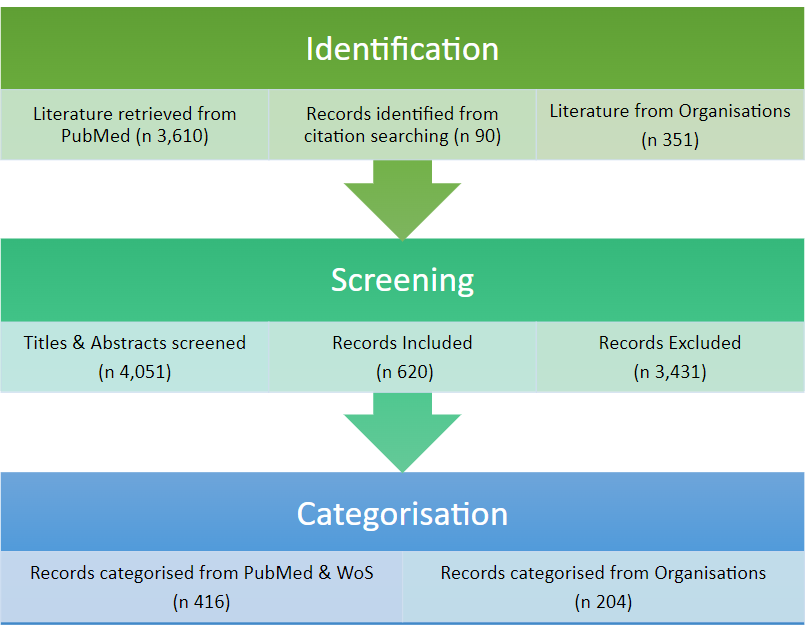
The final literature search results and their categorisation were reported in an excel document. The overall search strategy yielded a total of 620 unique records. The flow diagram for the process of reference selection is provided below (Figure 1).

The number of hits shown in Figure 1 indicates that the topic of NAMs is very broad. It covers digital advancements, “wet” biology, advanced and combined techniques and methodological discussions across all aspects of human toxicology. Even though clinical and environmental methods were excluded from the search, thousands of hits needed to be screened.
Despite the use of a third person to decide on the inclusion or exclusion of borderline papers, there is still subjectivity at play in deciding on the relevance of search results and in the classification of the identified literature, largely because the range and volume of the hits identified was so large. This meant it was not possible to assess the quality of the results reported in the studies included in the database other than for relevance to the goals of this project. As many papers detail opinions and perspectives from individual groups of experts or from conferences and workshops, traditional quality assessments for toxicological results do not easily apply so it is assumed that the peer review process for academic journals will ensure high quality literature (NB this might not apply to grey literature). Papers identified as being most relevant to the project are those that are discussed in the following sections of this report.
3. Expert stakeholder perspectives on the adoption of NAMs into regulations
The use of animal studies to understand the toxicology of industrial chemicals has historically been viewed as a necessary evil in the absence of a superior approach. The integration of non-animal methods into regulatory decision-making has been a goal for over 20 years, with a NAM-based approach to understand systemic toxicity proposed by the US National Academy of Science in 2007 (National Research Council, 2007). Transitioning from animal-based hazard-driven regulatory frameworks to a risk-based approach aligned with the results from NAMs faces numerous barriers along the way. The barriers can be technical around understanding the limitations of what individual or combined NAMs can provide, as well as institutional resistance rooted in a preference for established methods supported by legal precedence
One of the stated goals of this project was to “Summarise the expert opinions of the scientific needs still present which prevent further adoption of NAMs into the regulatory process. The focus here should document the current evidence gaps that need to be addressed but also any issues at a regulatory level affecting the confidence in interpreting NAMs data as regard decision making”. This has been achieved by a combination of interviews with selected stakeholders and a review of the literature published on this matter in the last 10 years.
3.1. Stakeholder Interviews: Methodology
The stakeholders were selected from a range of roles, including regulators, researchers, and industry. They were categorised according to their principal role but many also act as consultants and advisors to industry and regulators. The term regulator includes governmental or authorised organisations responsible for enforcing and/or co-ordinating chemical compliance. To maintain confidentiality, the identities of the interviewees are not stated in this report. The stakeholders were given a preview of the questions and were asked to complete the metadata questions before the meeting. Each interview lasted 30 – 60 minutes and were allowed to expand beyond the original questions so the interviewee could discuss areas of particular interest and experience. The following questions were asked to provide the metadata for the interviews.
Some interviewees stated they had dual roles that covered more than one category, so they were assigned to the one that best described the majority of their activities. Where an interviewee was active across multiple geographical areas or worked for an international organisation, they were assigned to the region where they worked for a majority of the time. Most interviewees stated they had either extensive or over 20 years experience in toxicology. One interviewee was a specialist in evidence methodology and computational research methods.
After consultation with the UK FSA, the following questions were used to guide the interviews:
Question 1: The acronym NAM is starting to be used to represent a variety of terms (New Approach Methodology; Non Animal Methods etc). Which is your preferred meaning for NAMs going forward or do you believe a different term should be adopted?
Although this question was intended to be a simple lead into the interview, we found that interviewees could express useful opinions on the fundamental aspects of NAMs without feeling the need to go into specifics. The basis of the question arose from views expressed by attendees to the European Commission organised workshop on the Commission roadmap towards phasing out animal testing for chemical safety assessments held on 11 - 12th December 2023 (EC, 2023).
Question 2: Could you comment on the amount of investment into NAM research and adoption into regulations in your region? More specifically on where it is being directed and how this is likely to change in the future?
This question was intended to understand the commitment that different regions have to NAM integration. We expected that many interviewers be able to give quantitative answers, so the stakeholders were able to comment on changes to investment instead.
Question 3: There appear to be contrasting perspectives in the community on the likely role of NAMs within regulations, either to supplement existing approaches or to completely replace animal testing. Which do you believe is the most likely direction in the next 5 years? Will this change in the longer term?
This question was intended to understand exactly how NAMs are likely to be integrated into the overall regulatory framework. It also allowed the stakeholders to express their opinions on the development of NGRA that could result in a paradigm change in how regulations are structured.
Question 4: Regulators use toxicology data for a range of reasons, such as prioritising regulatory activities, identifying emerging issues, setting safety limits and classifying substances. Do you think NAMs are more likely to play a role for one reason ahead of the others? Do you feel there are any aspects of the use of NAMs for food safety that need to be considered that differ from other types of chemical exposure?
This question is related to the previous one but allowed the stakeholder to go into more detail around which tasks regulations address that NAMs are most likely to be used in the short term. It also allowed the stakeholder to give their specific perspective around food safety.
Question 5: Difficulties in validation and regulatory acceptance are frequently identified as barriers to NAM adoption into regulatory decision-making. Are there other barriers to adoption or building greater confidence that need to be overcome also?
Although the interviewee was given the opportunity to discuss immediate issues around validation and acceptance, these have been discussed in great detail by other interviews, conferences and reports. The solution to the validation issue is often stated to be a need for greater funding, so we wanted to identify whether there are other barriers that could delay NAM integration that would mean future funding of validation would be less effective than hoped.
Question 6. Are there any emerging issues (e.g. mixtures, polymers, advanced materials) in chemical risk assessment to which NAMs would be well suited to address?
There are several emerging issues that align with chemical risk assessment, but the materials in question do not fit easily into the requirements for a simple mono-constituent substance. Such issues could include mixtures, polymers and microplastics, nanomaterials and advanced materials. It has been suggested that existing animal studies may need to be amended to address the physicochemical characteristics of these materials, so it could be regarded that the technical readiness level for NAMs is a lot closer to that of animal studies than it is for basic chemicals and hence integration might meet less resistance.
3.2 Key findings from the stakeholder interviews
3.2.1. Opinions on the use of the acronym NAM
Although there was a preference across the interviewees for the acronym NAM to represent ‘New Approach Methodologies’, several concerns and uncertainties were expressed. One interviewee expressed concern that “New” was a subjective term (UK, researcher) and another highlighted that some methods commonly included in the term are now over 20 years old. Several interviewees were concerned that “Non-Animal Methods” was too restrictive and could exclude the use of existing animal data or some new methods (e.g. unprotected animals such as invertebrates, early life stages, ex vivo methods using animal tissue), but others felt it shows the anticipated destination towards eliminating animal testing more clearly. Inconsistencies in the definition of “animals” in different jurisdictions was stated as being a barrier to the use of “non-animal methods”. “New Approach Methodologies” was felt to address all 3R’s (replacement, refinement and reduction), whereas “Non-Animal Methods” placed the emphasis on reduction only (NB the European Commission (EC) Directorate-General for Environment (DG ENV) sees the use of NAMs in refining rather than replacing animal testing). An EU regulator made the comment that for inclusion of NAMs into EU REACH (Registration, Evaluation, Authorisation and Restriction of Chemicals (Regulation (EC) No 1907/2006)) Annex XI (General rules for adaptation of the standard testing regime set out in Annexes VII to X ), a legal definition would be required. Whilst some interviewees felt the absence of a formal definition and a lack of consistency in its use impacts upon clear communication across the field because people have different understanding of the term (EU, regulator; Asia, regulator), agreeing on a definition was also perceived to be potentially very time consuming and difficult (EU, regulator). Many smaller regions are waiting to see the direction that the EU and US will take before making a commitment themselves (Asia, researcher). If different regulations/agencies decide upon different definitions, barriers in international acceptance of NAM data might be impacted.
The variety of different methods that can be covered by the term NAM was brought up by several interviewees, with one referring to it as a “galaxy of NAMs” (EU, regulator). To avoid misunderstandings, a UK regulator proposed that stakeholders should rather refer to the name or type of the method with more granularity where possible. Others suggested that, while NAM was very useful as a rallying point to give visibility to the overall topic (e.g. communication with non-experts, funding), it should not be used for more in depth assessments (UK, researcher; EU, regulator).
3.2.2. Views on the scale and direction of investment into NAM research
As most interviewees were based in Europe and North America, the opinions expressed focused largely in these regions. The difference in cultural perspective between regions was given as a future concern regarding the goal of NAM integration in regulations (i.e. would it be to improve safety for humans or for reduction of the use of animals) (Asia, regulator). There was a common consensus that these regions will drive investment so this focus was regarded as acceptable for this informal survey of opinions.
No interviewee was able to give a precise figure for the level of investment but all agreed that there is an upwards trajectory. The EU has committed over 1 billion Euros in the past years and investment is not expected to decrease (EU, regulator). Some large projects were specifically mentioned including: NIH project grants ranged from 3-5 million Dollars for data infrastructure development to 500 million Dollars to align in vitro and in vivo methods; ASPIS cluster (EUR 60 million); Partnership for the Assessment of Risks from Chemicals (PARC) (EUR 400 million). Reference was made to a paper by Carmichael et al. (2022) that details recent large projects in the area (UK, industry). Several interviewees highlighted that funding for research can come from many different sources and not always from those that specifically mention NAMs. This might explain why there were different views on the funding from the UK, with some interviewees stating it was negligible whereas others gave specific examples of funding in the UK. A second area where funding was felt to be lacking by several interviewees was around the validation of studies, especially the need for work with the OECD to be done on a voluntary basis. The importance of OECD work beyond validation was emphasised with the sandbox for IATA given as an example (EU, regulator).
There was a divergence in opinion regarding the approach of the US towards the integration of NAMs. Several interviewees cited specific examples of projects commissioned within the US, such as the US Food and Drug Administration (FDA) Modernization Act 2.0. This bill authorises the use of certain alternatives to animal testing, such as cell-based assays and computer models, to obtain an exemption from the FDA for investigating a drug’s safety and effectiveness prior to clinical trials. Additionally, the bill removes the requirement for animal studies in the process of obtaining a license for a biological product that is biosimilar or interchangeable with another biological product, allowing other approaches to be used to demonstrate similarity in safety, purity and potency (S.5002 — 117th Congress (2021-2022). Response to this bill emphasises that, although being welcomed as recognising the potential for these technologies, it is expected that “companies can now come to the FDA with alternative data as a starting point, but the FDA will continue to ask for animal data in almost all cases because that data is frequently pivotal to safe extrapolation into patients” (Henry Friedman, MD, deputy director of The Preston Robert Tisch Brain Tumor Center at Duke University in Durham, NC, and chair of the Foundation for Biomedical Research, quoted in Animal alternatives OK’d by new law (2023). Other examples cited were the ToxCast/Tox21 database and large scale government funded projects by the National Institutes of Health (NIH) National Center for Advancing Translational Sciences (NCATS) grants. However, another interviewee expressed a viewpoint that the US tends to be more reliant on animal data and may have limited awareness of certain organ models (UK, researcher).
Many interviewees pointed out that there was also heavy private investment, particularly within the pharmaceutical and consumer products sectors (a non-exhaustive list of active companies mentioned in the interviews were Unilever, Nestle, BASF, Syngenta, Emulate, Inspiro Medical). Although the usefulness of private research was widely accepted, it was recognised that the goal of these NAMs can be towards internal product development rather than regulatory risk assessment, so although increased transparency was regarded as a key goal the context of use would need to be considered when using any results. One area that it was felt that the pharmaceutical sector could provide important support was around the use of PBK models as they have been in use in this sector for over 20 years (UK, researcher). It was felt that industry seems willing to collaborate on public projects as only the results are used and not the intellectual property (EU, industry). Some projects that demonstrated this collaboration were stated to be the QI (Quality Improvement) initiative and the International Consortium for Innovation and Quality in Pharmaceutical Development - Drug Induced Liver Injury (IQ-DILI)) (N.America, researcher).
Outside Europe and the US, interviewees stated that whilst there was academic interest in NAMs with active research in both Japan and Korea (investment from KFDA (Ministry of Food and Drug Safety of the Republic of Korea)), it is on a smaller scale than in Europe and North America and often is done in collaboration with these regions (researchers and regulators, Asia, Australasia and N. America). Some areas do not have a coordinated push for the development of NAMs for food regulation (Australasia, regulator) and are expected to wait to see the direction that other countries will take (Asia, researcher). A UK based researcher pointed out that there is significant interest in India and Japan and a definite increase in funding for NAMs.
3.2.3. Perspectives on which NAMs are the focus of investment into research
Globally, NAMs are seen as attractive for investments in industry and research since there is more room for development and innovation than for existing methods. This may be more applicable to some sectors such as pharma, food and agrochemicals than others. Industrial chemicals manufacturers may have less incentive to invest into NAMs since the regulations that apply to them are often viewed as inflexible and require specified animal study data (UK, researcher).
Current investments focus on discovery and development of NAMs. Researchers call for more investments into complex endpoints such as developmental, reproductive and repeated dose toxicity. One technology that stands out is MPS (microphysiological systems). MPS have been used in pharma for the past 30 years (pre screening of drugs with known target substance and exposure), but to toxicology, the concept is relatively new. MPS offer a higher level of complexity in biological models than for example AOPs, and higher processing power (machine learning). There are some grants from NCATS (NIH) for MPS looking at neuromuscular junctions on a chip and comparing these to rat studies (N America, regulator). Other promising technologies are omics, 3D cell arrays, lung and gut models, and, recently, NAMs that deploy artificial intelligence (AI) and large language models. There is much less development on gut models than lung models, but gut models show better potential for particle toxicity prediction (M-cell, Peyer’s patch) (UK, researcher). In Germany some projects are ongoing related to organ on a chip and combining several organ models to present more relevant systems for food risk assessment. Other projects in the veterinary group are looking at emulating the gut on a chip (N America, regulator). A movement towards “hyphenated” methods was stated as an important advancement, especially where advancement in the sensitivity of analytical techniques will make some high throughput screening techniques more useful (n.b. the term “hyphenated” was used by the interviewee and was used to describe the linking of two methods within one experiment, for example linking gas chromatography with mass spectrometry to be able to both quantify and identify different components in a mixture) (UK, researcher)
In addition to the development of new technologies/methods, experts call for more investment into validation of NAMs, building infrastructure to share reusable data, and communication and training on the use of NAMs. There are different opinions on the structure the funded projects should have. While small and more targeted projects could be more appropriate (EU, regulator), broad, multi-disciplinary and multi-stakeholder projects could increase acceptance of the results (UK, industry).
Especially with the use of AI and machine learning, in silico NAMs have the potential to quickly generate large amounts of data which means that developing appropriate data infrastructure will be crucial to reuse high-throughput screening information for quantitative assessment (UK, researcher).
3.2.4. Current approaches towards the integration of NAMs into regulatory use
Looking at the implementation of NAMs into different national regulations, it is recognised that there is a divergence in the way NAMs are used which largely depends on the philosophy of the regulatory system (context of use). An EU regulator confirmed that the US risk based approach aligns with a greater acceptance of NAMs. There were conflicting viewpoints from the interviewees regarding the US commitment to NAMs with some highlighting their extensive use in screening and prioritisation whereas others mentioned an increased requirement to commission new animal studies in some areas whilst referring to them as NAMs. The EU uses a more rigid hazard based approach which is designed to use animal data. There also seems to be a slightly different focus on NAM usage where the EU are looking at speeding up assessment and the US at reducing animal usage. In the US and Canada, the burden of proof is on the regulators and they are willing to accept NAMs as long as supporting information is provided so that the methodology (its scope and limitations) are clearly understood. Hence NAMs are used, for example, in prioritisation for the assessment of existing substances and as rapid screen tests for food regulation as the current animal studies are too slow. Australia and New Zealand have a common approach across both countries only in food regulation and there is little coordination to push for NAM implementation. Use of NAMs is more prevalent in other areas such as veterinary medicines and pesticides where regulation in AUS/NZ follows the World Health Organisation (WHO), the Joint FAO (Food and Agriculture Organisation)/WHO Expert Committee on Food Additives (JECFA) and the FAO/WHO Joint Meeting on Pesticide Residues (JMPR) (Australasia, regulator). In Korea, hepatotoxicity in silico prediction finds application in support to in vivo studies, NAMs are generally used as screening to prioritise regulators’ work. Regarding food regulation, the KFDA is looking for NAMs in the prediction of toxicity of dietary supplements from natural products (mixtures) (Asia, researcher).
3.2.5. Comments on how NAM integration will be done in the future
When asked about the roles NAMs will play within the next 5-10 years, there is a consensus across the stakeholder groups that NAMs will have a great impact on current regulations in the long-term but they will not be able to completely replace existing in vivo studies for the foreseeable future. It should be noted that this response was inline with a survey commissioned by the European Chemicals Agency (ECHA) that stated of (134 respondents), 40% saw full replacement of animal studies with NAMs in industrial chemical assessment within next 10-20 years and 27% expected it to take over 20 years. A few interviewees gave perspectives that there were opportunities to reduce the quantity of animals used in testing.
-
Advances in large language models and artificial intelligence have only been significant in the last couple of years and these could accelerate the use of NAMs (UK, researcher)
-
Integration of legacy animal data with NAMs should allow animal studies to be done on completely new chemicals of which there will be a limited number (UK, industry)
-
NAMs will allow targeted testing rather than a fixed array of tests in 5 - 10 years reducing animal use by 50 % (EU, regulator)
One learning over the past years has been that the 1:1 replacement of traditional testing with NAMs is not considered appropriate anymore and that multiple NAMs will be required depending on the complexity of the endpoint. Several interviewees discussed example where NAMs were already integrated, including OECD test guideline (TG) 497: Defined Approaches on Skin Sensitisation, skin irritation and corrosion, eye irritation and damage, and the use of TTC and QSAR for pesticide and biocide metabolites (UK, industry). According to an EU regulator, using a combination of in vitro studies and short-term in vivo studies has been applied to reduce the use of animals to assess endocrine disrupting properties. Similar strategies are being considered for cancerogenicity and neurotoxicity testing (UK industry). Approaches to combine different NAMs and integrate the data were regarded as vital to the transition towards NAMs usage (UK, researcher). However, the current approach to validation would not be viable for batteries of studies (UK, researcher)
There is great potential for in silico, in vitro and omics data to support existing methods, chemical grouping and RxA approaches, as well as in multi-tiered screening processes, ranking and regulatory prioritisation of chemicals. Especially with the use of AI, in silico NAMs can quickly generate large amounts of data and could be used early on in the assessment process to quickly gather hazard and exposure information, e.g for pesticides, food contact materials, food additives, naturally occurring substances (e.g. plant alkaloids), contaminants and even complex mixtures or substances difficult to test in vivo (e.g. nanomaterials). NAMs can be integrated into TTC approaches and hypothesis driven IATAs. Grouping and read-across are currently used for industrial chemicals (e.g. Per- and polyfluoroalkyl substances (PFAS)), food additives (US) and even enzymes in an approach called ‘safe strain lineage’ (Australasia, regulator).
Within food approvals in the US, there has been a move away from dog studies using NAMs for rapid screening and mechanistic insight into substances of concern, since the burden of proof is on the regulators and current animal studies take too long (N. America, regulator). In contrast, a Japanese regulator seemed more conservative, pointing out that the current in vitro and in silico NAMs have significant limitations in terms of long-term ingestion and the assessment of food additives in mixtures.
Researchers in both academia and industry see potential for NAMs in the short-term to fill gaps of traditional methods and for little developed endpoints such as hepatotoxicity (drug induced liver injury, DILI), neurotoxicity and developmental toxicity. This may be especially interesting for the more hazard based risk approach in the EU where authorities demand more data on potential toxicological effects, such as developmental and neurological effects linked to Parkinson’s disease and obesity.
3.2.6. Regulatory activities where NAMs can play a key role into the future
The current regulatory activities where NAMs were expected to play a role in the immediate future were largely around screening, prioritisation and ranking, grouping and RxA. Very few interviewees were of the opinion that NAMs would be used for quantitative regulatory decisions such as setting exposure limits, classification or placing bans/restrictions on the use of chemicals without being combined with in vivo data. One area that was proposed as an exception to this was where rapid decisions were needed around substances with little available data. A couple of interviewees placed different NAMs within a tiered testing strategy that included animal data on at least one other stage, with in silico data being used at Tier 0 (i.e. screening) (EU, regulator) and omics data at Tier 3 (i.e. to explain observed toxicity) (UK, researcher). Concerns over reductions in human health safety from the rapid transition to NAMs were expressed, especially the potential for false negatives, with a recommendation that their use should only be used for interpolation not extrapolation (Asia, regulator).
Regarding their use for food safety, several interviewees expressed an opinion that NAMs could be used in this sector ahead of others. The reasons given for this were that food safety has less regulations where prescribed tests are needed and that the substances under examination may have little data. Examples of cases where NAMs have already been used for food safety assessments were given:
-
NAMs to support grouping and RxA of microbial consortia (North America, regulator)
-
Grouping and ranking of alkaloids for further testing (UK, researcher)
-
Prediction of toxicity of dietary supplements from natural products (Asia, researcher). It was noted that although information on mixture toxicity was requested, it could only be provided for single substances. The difficulty of predicting mixture toxicity was also reported by a Japanese regulator
The relative lack of gut models, particularly those that include the gut biome, when compared to other routes of exposure was emphasised (UK, researcher). Another UK researcher stated that regulators need to define the context of use of NAMs to ensure they were fit-for-purpose. Finally, the difficulty in using NAMs to predict “low toxicity” was discussed (EU, regulator). The interviewee used the term “low toxicity” to recognise that all substances are toxic if the dose is sufficient, but many do not induce an adverse effect at a dose that can be expected in normal circumstances. This means the term “a non-toxic substance” is viewed by some as scientifically meaningless. In the EU and UK, substances that do not trigger an adverse effect at a realistic dose are defined as non-hazardous. The issues around using NAMs to predict “low toxicity” are discussed in more detail in Section 4.6.
3.2.7. Barriers to NAM integration into regulations
Validation
Although a request for barriers other than validation were requested, it became a key discussion point in most interviews. The general opinion was that the existing validation process in the OECD is long, convoluted and prone to political influence (UK, researcher). As the methods being validated are being used for health protection, a comprehensive approach was regarded as essential but the existing approach was viewed as out of date (guidance is over 20 years old but is being updated currently) and not applicable to fast developing technologies such as NAMs (UK, researcher). Key mechanisms to build confidence in using NAMs will be the establishment of validation panels, education and transparency from regulators as exemplified by the 2023 EU Reference Laboratory for alternatives to animal testing (EURL ECVAM) status report and case studies. In principle, test methods and approaches could also be validated by other actors than validation bodies. For example, metabolomics best-practice and reporting guidelines for regulatory toxicology were published by the MEtabolomics standaRds Initiative in Toxicology (MERIT) consortium (Viant et al., 2019) and used in a ring-trial approach to assess metabolomics reproducibility for chemical grouping (Viant et al., 2024). However, the impression prevails that EU regulators will heavily rely on validation of in vitro/NAMs by in vivo data for the foreseeable future. Interestingly, validation is not as critical for the US FDA in reference to NAMs. Previously, expensive ring-trails were needed, however, these are now replaced by “qualification” which takes into consideration the context of use (N. America, regulator). For example, for a screening NAM, the consequences are limited, so limited qualification would be needed for justification of the results. The US Interagency Coordinating Committee on the Validation of Alternative Methods (ICCVAM) is starting to use this approach. Also, the US FDA ISTAND (Innovative Science and Technology Approaches for New Drugs) program is looking to promote qualification of methods that do not fall under the remit of existing qualification programmes. Another term expressed by an EU regulator was “endorse”, primarily associated with complex approaches such as AOPs and IATA. A more technical concern around validation of new methods was how to identify appropriate positive and negative controls against which to benchmark a validation (UK, industry)
Beyond validation
A commonly expressed view was that a lack of confidence and trust in NAMs from regulators was a significant barrier. This was attributed to a preference to work with established approaches (EU, regulator) but it was also highlighted that decisions made by regulators directly impacted people’s health so until NAMs can be shown to be an improvement on existing approaches they should not be used for decision-making (Asia, regulator). An interesting perspective was that most regulations need to use “the best science” and in some cases NAMs should be regarded as meeting this requirement (e.g. the defined approach for skin sensitisation was understood to be more predictive than animal studies) (UK, industry). Where some interviewees suggested that training should overcome this (EU, regulators; UK, industry; North America; regulator), others felt that there was a more fundamental cultural conservatism that will need a new generation of regulators (UK, researcher). Adding to the slow progress is a lack of flexibility in some regulations, especially those with detailed requirements and defined methodologies such as EU REACH for industrial chemicals. The key study approach will be difficult to apply considering a multitude of NAMs is needed to fulfil one endpoint. According to EU regulators, there has been some movement to change CLP to align with NAMs but it is difficult as the classification system relies heavily on animal data. NAMs could more easily be used where there is no defined methodology described within the regulation so as for food additives (EU researchers). Another point highlighted by an EU researcher was that in silico NAMs used in lower tier screening might not necessarily have to be fully optimised for reaching an immediate, short-term goal or an approximation. This importance of understanding the context of use was emphasised by other interviewees (UK, researcher). Interviewees from the UK industry stated that education and training resources were becoming available from the Animal-Free Safety Assessment (AFSA) initiative, and the International Collaboration on Cosmetics Safety (ICCS).
NAM integration is also a social, cultural and economical issue. Unfamiliarity can lead to resistance to change. The public opinion will be shaped by questions of safety and the level of understanding of NAMs but will also depend on the application of the assessed substance. It was felt that consumers are more accepting of animal testing in drug development and food (e.g. gene modified food) than for cosmetics (Australasia, regulator). On the other hand, the public can apply pressure on regulators to reduce animal testing, as for example by the EU citizen initiative pushing for investments into non-animal methods (UK, researcher). An interviewee from UK industry commented that the PARC project will examine this but the project has not yet reported on this topic.
Several technical barriers beyond those previously mentioned were discussed. Although NAMs used to represent models are complex they still do not represent the complexity of an organ itself. For example, there are very few NAMs that account for blood flow within capillaries. Models to represent the gut are challenged by the requirement for both anaerobic and aerobic environments in the same study and microbiome viability is short (2 days) (UK, researcher). One interviewee expressed that there was a lack of suitable data sharing infrastructure in place meaning it is very expensive to find, access and reuse data and integrate it in another system. They highlighted that the US NIH (National Institute of Health) is awarding grants for developing data infrastructure (3-5 million dollar) and that the EU should look to match this investment (UK, researcher). Other economic barriers were also identified. It was thought that a battery of NAMs would be more expensive than a single animal study, with the example of the comparative cost of the Defined Approach for skin sensitisation and the animal study it replaced being given (UK, researcher). A UK-based interviewee from industry stated that if there was no regulatory adoption of NAMs there was no incentive for CROs to invest in providing NAMs.
3.2.8. Emerging substances and materials where NAMs are well suited to play a part in their risk assessment
Interviewees gave a wide range of different substances and materials where NAMs could play a prominent role in their risk assessment. Many of these were substances where there was no precedence of testing using animal models, although it was emphasised that initially animal testing and NAMs would need to be done in parallel to ensure that the NAMs were performing as expected, and benchmarks and controls could be established. Several interviewees also identified materials where there could be many different compositions needing assessment, such as mixtures and polymers, where the quantity of animals to test every example would be simply too high. Grouping and RxA were suggested as a good approach to achieve this (UK, researcher) and omics were specified as being a NAM that would be well suited to justify this approach. One interviewee emphasised that the tendency of omics to produce false positive results would be ideal to screen for non-hazardous polymers (UK, researcher). An EU regulator also described how multi-omics could be used to assess complex new materials such as biopesticides, microbials, botanicals and double stranded RNA. They also suggested that NAMs would be well suited for substances that would not be available on large scale, such as impurities, metabolites or degradation products as animal studies can require multigram or even kilos of the test substance. One specific example mentioned by more than one interviewee was how NAMs could be used to better understand health concerns raised by 6PPD-quinone, a degradation product of an ingredient in tyres that is currently receiving much regulatory attention (N. America, regulator; EU, regulator). NAMs could also play a role where there are issues with sample preparation for animal studies (e.g. nanomaterials) (Asia, researcher) or where there are no established animal models (e.g. oral allergenicity) (Australasia, regulator).
Many interviewees reiterated that NAMs could only be used for these materials once they had been shown to fall in the applicability domain of the test. As demonstrating this for simple substances is not trivial, it was expected that it could be even harder for more complex materials (Asia, regulator; EU, regulator). A UK-based researcher discussed the specific challenges of preparing stable dispersions of advanced materials or polymers. Many NAMs are designed to use the test substance in an aqueous solution. Although tests can be adapted for insoluble substances, they require stable dispersions to facilitate biological uptake and for dosimetry to be understood. The difficulty in forming stable dispersions of nanomaterials has been recognised (Hartmann et al., 2015) and guidance produced by the OECD (2012). The interviewee was expressing their view that similar difficulties may be seen for some insoluble advanced materials and polymers.
4. Summary of literature search: Wider perspectives on NAMs and their integration into regulations
There has been a plethora of literature published giving opinions, recommendations and counter views around the integration of NAMs into regulatory frameworks. These have covered a wide range of topics from overarching perspectives on the rate of change in the regulatory paradigm to focused discussion of the position of individual NAMs in a specific regulation for a selected endpoint. A review of opinions expressed in published reports in academic literature and grey literature over the last 10 years is presented.
4.1. Criticisms of the existing regulatory toxicology paradigm
For there to be a change in the accepted way to perform tasks there needs either to be evidence that a new way is superior or the existing approach is not performing as required, and this applies as much to regulatory toxicology as it does to other processes. Many advocates of the rapid integration of NAMs into regulations state that animal studies are regarded as “a gold standard”, but the drawbacks of using animal studies, both ethical and scientific, have been widely discussed. The 3R’s principle (Replacement, Reduction and Refinement was originally developed in 1959 in The Principles of Humane Experimental Technique (Russell & Burch, 1959) requiring an adequate ethical consideration of animal welfare during research and has become an integral part of much transnational legislation (Grimm et al., 2023)). Despite this, the bedrock of most chemical regulations aimed to reduce risk to human health is the use of animal studies to identify and quantify the hazard of chemicals, based on the view that observation of adverse effects in animals is the best available predictor of similar effects in humans (D. J. Knight et al., 2021). However, there is increasing evidence that animal studies cannot be regarded as infallible, particularly from the pharmaceutical sector where toxicology to animals and to humans can be directly compared (Macmillan et al., 2024).
-
Systematic review indicated that 18 % of animal studies were contradicted by human randomised trials (Hackam & Redelmeier, 2006)
-
Examination of six medical interventions showed only 50 % concordance between animal studies and clinical observations (Perel et al., 2007)
-
Animal studies have failed to predict severe toxicity in humans during pharmaceutical development (Olson et al., 2000; Van Norman, 2019)
-
Only 8 % of Active Pharmaceutical Ingredients (API) that enter Phase I clinical trial gain approval (Harding, 2004)
In addition, the existing validated animal studies commonly required in regulations do not address some common human health outcomes such as developmental neurotoxicity, neurological diseases (e.g. Parkinson’s) and endocrine disorders (e.g. endometriosis, type II diabetes) (Bennekou, 2019). Hilton et al., 2023 have described how paradigm shifts in the understanding of science occur by the accumulation of ‘anomalies’ to the accepted science until ‘crisis’ requires a replacement of the beliefs in the accepted science. They postulate that regulatory toxicology is in this situation currently with several regulations based on the science of the late 20th century (e.g. the Federal Insecticide, Fungicide, and Rodenticide Act in the US has not been amended to update toxicity data requirements since the Food Quality Protection Act of 1996). Leist et al., 2014 has listed a series of specific technical issues around animal models: High false positive and negative rates; lack of validation; low predictivity; frequent waiving; uncertain high dose-low dose extrapolation; metabolism and biology different to humans.
The call for a paradigm shift in regulatory toxicology is increasingly being expressed in academic literature (Berridge et al., 2024; Burden et al., 2017; Fentem et al., 2021; Hilton et al., 2023; Johnson et al., 2022; Punt et al., 2020). Beyond the fallibility of the predictive performance of animal studies, the relevance of some of the exposures required by validated animal studies to those received by humans has been called into question (Gellatly & Sewell, 2019a), a perspective confirmed by an interviewee in this project. Although using high doses is a conservative approach, in-line with the precautionary principle, fears have been expressed that the doses are not relevant because they cause excessive toxicity or saturate metabolic pathways (ECETOC, 2021).
4.2. If not animal studies, then what?
Very few of the proponents of a paradigm shift away from animal studies for risk assessment suggest that a single NAM can act as a 1:1 replacement for an animal study and this view was confirmed by the stakeholder interviews in the previous section. This is primarily due to the basic difference in the goals of the two types of approach. Animal studies have been described as an observational approach, whereby animals are dosed with a chemical, often at levels much higher than would be expected during normal usage, and the animals observed to see what effects this has on them. Although some studies include sampling of blood and tissues for analysis, there is little information generated as to why the observed effect has occurred (N. Ball et al., 2022). Several regulations in the EU and other regions use this observational approach associated with a top-down hazard based assessment to underpin their structure. This method prioritises identifying the doses at which toxic effects occur and do not occur, over understanding the underlying mechanisms of these effects (Table 3).
Understanding why an adverse effect can be important for specific aspects of the regulations, for example:
-
Whether the reason for a substance being placed on Annex XIV of REACH is a threshold or non-threshold effect defines the approaches available to apply for authorisation
-
To justify the use of an adaptation under Annex XI of REACH to use a WoEapproach or read-across instead of generating new data
-
If a substance is evaluated and concerns over it being an endocrine disruptor are raised, the registrant may be required to perform studies that examine the mechanism of an adverse effect, but this would only be needed for a small number of substances
-
To justify the use of bridging principles to classify a substance or mixture under CLP
An alternative paradigm that focuses on the extensive use of NAMs suggested by many stakeholders is based on a predictive, scientific approach that is bottom-up and risk based. It uses detailed understanding of the biological mechanism leading to toxicity to explain or predict the adverse effect at a given dose or to justify the use of existing data to address the endpoint. This data needed to understand the mechanism is expected to be largely derived from NAMs and thus minimise or even remove the need to commission new animal studies. Due to the complexity of biological mechanisms and the design of many NAMs, in vitro studies especially, examining a specific aspect of the mechanism only it is expected that a battery of NAMs will be needed to give the holistic perspective that an animal study gives. Many stakeholders do emphasise that should the battery of NAMs give a comprehensive perspective, it will be of the toxicity to humans not to a different mammalian species, so should be regarded as superior. This opinion extends to discussions on the overall goal of NAMs and how they should be validated in the future. By using data from animal studies as a benchmark against which the performance of a NAM or battery of NAMs is assessed, the NAMs are expected to be predictive of toxicity to that animal. Some commentators believe that this is not the ultimate goal of regulatory toxicology and that this is the protection of human health. Middleton et al. (2022) advocate for this approach in NGRA, choosing the NAMs by their ability to detect biological perturbations at low exposures, use toxicokinetic modelling to predict the exposure where this effect might be triggered and compare that with predicted exposure.
Whether taking a predictive or protective approach to risk assessment, the current predominant approach to establishing a scientifically justified battery of NAMs is by using the AOP approach.
4.3. Considerations on the strengths and weaknesses of the AOP approach
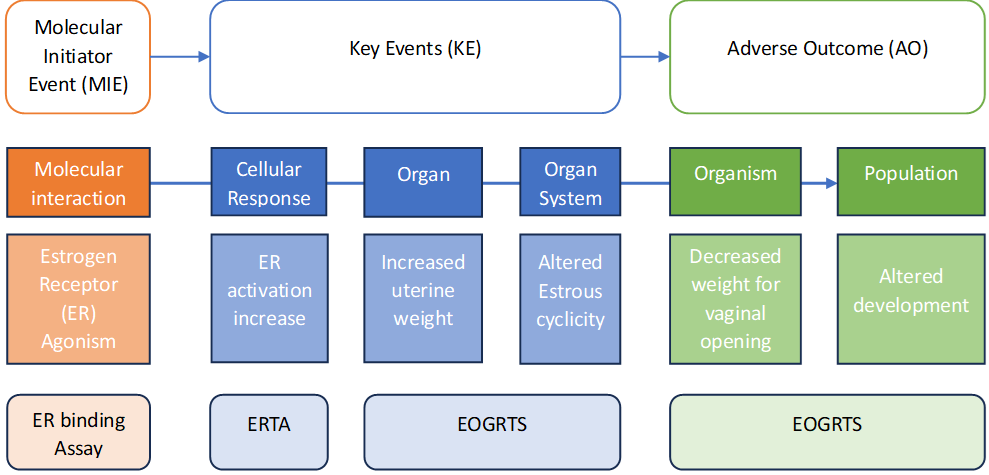
The AOP approach to understanding toxicity breaks the biological pathway that leads to an adverse effect in an organism into individual steps from an initial exposure event through a series of key events (KE) culminating in a relevant adverse outcome. Where an AOP is well understood it is hoped that knowledge of the earlier steps in the AOP would result in confident prediction of the effect at an organism and population level without the need to directly test on the animals. The AOP approach provides a structured, hypothesis driven alternative to relying on animal studies that is based on an understanding of mechanistic effects instead of observation. It allows the integration of data from different levels of biological organisation, gives a scientific basis to the development of testing strategies and has the potential to give evidence to support the use of existing animal data to replace the need to commission new studies (Bajard et al., 2023). There is now an extensive online library of AOPs (AOP-Wiki, n.d.) with associated guidelines and there is a drive to standardise their description to allow the relationships between different AOPs to be more clearly understood (OECD, 2018b; Villeneuve et al., 2023).

There is significant overlap between the AOP concept and the Mode of Action (MoA) framework proposed by the International Programme on Chemical Safety (IPCS) in 2001 (Boobis et al., 2006), the key difference being that an AOP considers the interaction between biological mechanisms irrespective of the chemical that may initiate it, whereas on MoA would examine a comprehensive molecular description of every biological event in the pathway from an initial interaction with a specific chemical (Lima et al., 2021). Figure 2 exemplifies some of the strengths and weaknesses of the AOP approach. Understanding the MIEs and KEs are often best done using NAMs, in particular in vitro studies that are specifically designed to focus on the specific biological event and can be designed to be aimed towards toxicity to humans specifically, for example by use of human derived cell lines. However, understanding a KE at a higher level of complexity such as an organ is usually beyond the scope of a simple in vitro method alone, requiring either a battery of studies, organ-on-a-chip technology or more usually an in vivo study. Svingen (2022) states that as the complexity of a system increases, the more emergent properties arise which cannot immediately be predicted from knowledge of the basic mechanisms occurring in that system. He refers to a quote by Anderson et al. (1972) that states “The ability to reduce everything to simple fundamental laws does not imply the ability to start from those laws and reconstruct the Universe”. This can be explained in part because an AOP does not exist in isolation as a linear sequence of events in a living organism, instead it is a part of a network of mechanisms that impact on each other (T. Ball et al., 2021). As the AOP approach is agnostic of the chemical being assessed (unlike the MoA framework), it does not account for the toxicokinetics and toxicodynamics of the chemical in question, meaning that an AOP in isolation might not be sufficient to properly predict the dose at which point an adverse effect might be observed, which is necessary for many regulations. Bajard et al. (2023) described the limitations on AOPs.
-
Non-standardised ontology limits the use of machine reading technology that would significantly speed up the development and standardisation of AOPs.
-
Incomplete information across the AOP. Even if a KE is postulated there may be limited information to confirm its relevance
-
Slow reviewing and endorsement of AOPs
-
Predictable dose-response relationships are rare, making quantitative risk assessment based on AOPs difficult
These perceived weaknesses have led to several researchers suggesting ways to evolve the AOP approach. Ball et al. (2021) discuss the situation where a single KE may be part of a series of different AOPs, so the researcher needs to know not only the correct assay with which to assess the KE but also what the next steps should be given the result of this assay, for example which adverse outcomes should be a concern and which follow up assays will give the most useful information. This means that careful choice of any initial screening studies can give information on many different AOPs, something that would be essential for the use of NAMs in the more complex regulatory endpoints such as repeated dose toxicity, for which animal studies can provide observation information arising from many different NAMs across many different organs, whether they are intended to supplement or to replace the animal studies. The hypothesis driven, step-wise nature of AOPs mean that they align well with testing strategies that show a similar structure, such as IATA and Defined Approach (DA). During the stakeholder interviews, one interviewee (UK, academic) also highlighted that the AOP approach was a simplistic view of the complex biology and that new technologies in both microphysiological systems (MPS, includes organ-on-a-chp, organoids and 3-D cultures with flow and microbiome capabilities (NC3Rs, n.d.), machine learning and artificial intelligence should allow the assessment of mechanisms that were previously too complex to model. As discussed in a separate section of this report, regulatory validation of these methods is not expected in the near future, so they will likely remain as supporting or screening technologies in the short-term. Omics were also suggested as a useful technology to support a transition from simple linear AOPs to a network of AOPs as they are able to screen a wide range of MIE and early KE at once.
4.4. Use of IATA and Defined Approaches (DA) in regulatory assessments
Many existing regulations are heavily prescribed with an obligation to assess stated endpoints with a limited number of studies and these are often animal studies. This approach is irrespective of the likelihood of an adverse effect being observed and with little consideration of adverse effects that would not be detected by these studies. The EU regulation REACH has been described as an example of this type of regulation leading to the accusation that it has required the use of too many unnecessary animal studies. Knight, Hartung and Rovida have estimated that 4.2 million animals have been used for REACH systemic toxicity studies (J. Knight et al., 2023). The use of NAMs to demonstrate an AOP have been suggested as an alternative approach and the use of IATA and DA are the approach by which the tests required to examine an AOP can be structured.
IATA are a pragmatic science-based approach that take an iterative approach to examine a hypothesis related to the toxicity of a substance. They have a three-part generic structure (Casati, 2018): (i) collect existing information; (ii) WoE evaluation; (iii) generate new information (if needed). They are intended to be flexible in nature and require expert input to both design the experiments and to interpret their results. This means that their use may be more appropriate for regulations where responsibility for safety falls on highly trained regulators, such as the US Toxic Substances Control Act (TSCA) regulations. Where responsibility rests on the manufacturers and importers, the use of a more explicit approach would reduce the risk of poor-quality data. It must be noted that regulations of this type, such as REACH, do have mechanisms whereby alternative approaches to satisfying regulatory endpoints do exist, such as grouping, RxA, WoE and data waiving based on exposure or physicochemical properties (REACH Annex XI).
Even where the alternative approaches are not applicable, the use of NAMs is not entirely precluded. Defined Approaches use a fixed set of information requirements and data information procedures to ensure consistency in application. They can be structured in a stepwise fashion allowing the testing to be discontinued partially through the process if a strong conclusion can be made before all the aspects of the DA are completed, but this decision would be made according to a set of pre-defined rules. Where the studies in a DA have been validated, they can be covered by an OECD Test Guideline and fall under mutual acceptance of data for OECD member countries. Casati et al. outlined criteria that all studies in a DA should meet (Casati et al., 2018). They highlight that IATA can be adaptable to the needs of a particular regional or sectorial regulatory requirement whereas DA would be applied in the same way across all jurisdictions. The use of NAMs in a DA has been demonstrated for the skin sensitisation endpoint in OECD Guidance Document (GD) 256 (OECD, 2017a), whereby AOP key events can be examined using different validated methods based on knowledge of the biological mechanisms leading to skin sensitisation.
Data integration
The use of risk assessment approaches that use multiple data sources such as AOPs IATA and DA requires methods for data integration. The first barrier to adequate data integration is poor and unorganised reporting of results from individual studies. Numerous templates and guidelines have been developed for the reporting of different types of study. Escher et al. (2022) give a comprehensive and in depth assessment of these tools and they are also listed in Annexes 4 and 5 of this report. Whilst frameworks to report (Q)SAR and PBK models are regarded as regulatorily accepted (e.g. GIVIMP, OECD TG 211), those for omics are generally in the research phase. One exception is the Best practice guideline on metabolomic by Viant et al. (2019) which was categorised as being under in situ validation.
Beyond the reporting of individual results, data integration requires results from different studies to be pulled together to reach an overall conclusion. Currently this is largely done by expert judgement leading to the potential for inconsistency and bias. One area of concern is how to collate and synthesise the uncertainty around a decision from the uncertainty associated with different studies. As these may be very different studies, comparing and contrasting them will need great care. This leads to two more difficulties that will be faced during data integration emphasised by Escher et al. (2022).
-
Absence of common ontologies across different methods
-
Absence of an overarching framework for data integration
They present a number of guidance documents designed to assist in data integration but none are more advanced than in situ validation regarding their regulatory readiness.
4.5. Context of Use
A term that was repeatedly brought up by stakeholders and appears in many publications on NAMs is the importance of the context of use of the NAM on its regulatory acceptance. This can be both in terms of the regulation within which it is being used, but also the role that it is playing in a specific assessment.
Regulators perform a wide range of tasks, which are defined by both the substances and uses that their remit covers and the region within which they operate. The role that NAMs can play for regulators depends on the context within which they are being considered. Cote et al. (2016) summarised this situation from a US perspective (Figure 3).

The authors expand on the characteristics of a “fit-for-purpose” assessment for each tier. They recommend that the use of in vivo data to address human variability moves from none in tier 1 to essential in tier 3 and that the consideration of metabolism in test systems moves from some to none for tier 1 to comprehensive in the intact organism for tier 3. Parish et al. (2020) present this discussion in a different context. They identify a series of criteria that define fitness-for-purpose and explain how the importance of each criterion can vary depending on the context of use (Table 4). The authors present case studies that assess different NAMs against the default criteria importance in different regulatory situations.
The context of use is defined by the requirements of the regulation within which a risk assessment is being done and can vary by region and/or sector of use. For example, many regulations in the US place the burden of responsibility on the relevant federal agency. As these agencies have limited resources, they cannot examine every substance placed on the market meaning they need to screen and prioritise the substances to identify those that require immediate attention. Screening and prioritisation does not require a final, fully scientifically justified conclusion to be drawn so there is more freedom in the tools that can be used for this. In a presentation to an Society for Chemical Hazard Communication (SCHC) meeting in 2018, Scarano (2018) summarised the EPA’s strategic plan (US EPA, 2018) to promote the development and implementation of alternative test methods within the TSCA Program as mandated in the amendment to this regulation in the Frank R. Lautenberg Chemical Safety for the 21st Century Act. He states that some NAMs have been used for a number of years in the new chemicals programme, particularly for chemical and hazard characterisation (e.g. EpiSuite, ECOSAR (Ecological Structure Activity Relationships), Oncologic). The EPA would consider NAMs for screening, prioritisation and chemical risk assessment. ICCVAM (2024) have collated reports that describe the testing needs for different topics and different federal agencies. They recognise that many NAMs are developed for the purposes of research, especially in the pharmaceutical sector, to assist product development decisions. Whilst these may not need to meet the quality requirements for regulatory use, the data they generate can still provide useful insights so the basic criteria of transparency and applicability should also be applied to these NAMs. This position is backed up by the responses to the stakeholder interviews, where the importance of the research by the private sector was acknowledged and efforts should be made to improve the inter-sector transferability of data and learning.
In some regulations, the EU takes a slightly different approach, whereby responsibility for risk assessment lies on the manufacturer or importer of a substance. For example, REACH requires that all manufacturers or importers of a substance on over 1 tonne per year provide a proscribed set of hazard data (content of which is tonnage dependent) and a risk assessment if the substance is classified as hazardous and more than 10 tonnes per annum is placed on the market in the EU. This obligation partly removes the need for the regulators to perform a screen from scratch. However, where a concern is noted from the data in a registration dossier, the regulators do use screening tools such as (Q)SAR and databases to expand the available data on which to make a decision on whether further information is needed from the registrants. This is evidenced by the use of findings from the Toxcast/Tox21 data bases in the Annex XV dossiers for 4,4’-(1-methylpropylidene)bisphenol (bisphenol B; BPB) and 4,4’-sulphonyldiphenol (Bisphenol S; BPS) (Belgium FPS Public Health, 2022; FR-MSCA, 2021). Other EU regulators do need to use screening and prioritisation, particularly where they need to assess the safety of substances that are not intentionally produced, such as regulators of food safety who would assess degradation products and metabolites.
4.6. Use of NAMs to identify low toxicity substances
Many regulations are structured to identify substances that present a hazard and risk to human health. Regulators will then implement measures to minimise the risk that such substances present. However, as there is a drive for innovators to build safety into the design of their chemical products, identification of “non-toxic” or low toxicity substances is perhaps as important as identifying toxic substances. It must be noted that the term “non-toxic” is misleading as toxicity is often observed if the target is exposed to enough of the substance (Sola dosis facit venenum [the dose makes the poison] - Paracelsus). A criticism of the existing approaches to regulatory toxicology using in vivo studies is that the doses used in the studies far exceeds those expected to be seen during expected use of a substance. An aspect of NGRA that advocates for a paradigm shift in toxicology highlights is that use of NAMs, particularly including the use of PBK models, can demonstrate “non-toxicity” at the dose that is expected to be received by humans. There is a subtle difference in the approach to identify “non-toxicity” and toxicity as the latter requires the proof of a positive result in studies whereas the former requires the evidence of a negative result. As animal studies can be regarded as observational, they can equally be used to observe both adverse effects and the absence of adverse effects in the entire animal. The use of a battery of NAMs used to examine a specific AOP is predicated on identifying the occurrence of a specific pathway to an adverse effect rather than the adverse effect itself. Where there are a limited number of well understood mechanisms to an adverse effect and there are validated NAMs to examine the key events in the mechanisms, the absence of toxicity can be as equally demonstrated as the presence of toxicity. However, the demonstration of “non-toxicity” for systemic endpoints could require the examination of multiple AOPs and there would always be the concern that knowledge of AOPs is incomplete and a biological mechanism towards an adverse effect has been missed. Rovida et al. (2021) discuss the difficulty in selecting the correct NAM to support read-across for low toxicity substances. Using an exhaustive approach to address all possible mechanisms could prove to be prohibitively resource intensive. The requirement to identify non-toxic substances must be included when designing the battery of NAMs for regulatory purposes. The topic was discussed in the ECHA workshop “NAMs in Regulatory Science Proceedings of a scientific workshop Helsinki” (2016c) and whilst it was acknowledged that NAMs can achieve this goal, it had not been a principle goal for much research so there was less data available to further explore the topic. Metabolomics were discussed as a specific NAM that could help to achieve this goal.
4.7. National and regional strategies to further integrate NAMs into regulations
In order to link the perspectives stated by the interviewees with the activities underway in different parts of the world, we include a list of different roadmaps, strategies and frameworks published by regulators identified by the literature search in Annex 5. Seven overarching roadmaps and strategies and work plans addressed the entirety of regulatory toxicology rather than concentrating on specific NAMs or endpoints, from UK (COT, 2021), EU (EFSA, 2022), US (EPA, 2018) and Canada (Bhuller et al., 2021). Examination of the main aspects of these documents reveal commonalities in goals across different geographical regions and with the UK FSA roadmap published in 2023.
-
Each roadmap was written on the assumption that greater integration and use of NAMs in regulations was desirable
-
All documents emphasised the need for confidence building in NAMs across stakeholders and outlined plans to achieve this goal
-
All stated the need for integration into the existing regulatory framework in the near term. Interestingly, only documents from US and Canada explicitly discussed regulatory flexibility and a paradigm shift in regulatory toxicology
-
A topic where the UK FSA gave a more detailed discussion on future plans was around the use of digital technology although all regions discussed the importance of data sharing and integration
-
The need for training of regulators was discussed in all documents but European Food Safety Authority (EFSA) also discussed the need for 2-way training to allow regulators to inform researchers about their needs, a theme that was raised in the stakeholder interviews several times
-
Although many different techniques are covered, the use of PBK models and IVIVE receive particular attention across most of the roadmaps
There were some details in these documents that were not prominent in the UK FSA roadmap and may be worth considering in the future.
-
EFSA included a detailed discussion on exposure (referred to as the exposome) assessment using human data such as biomarkers. The US EPA also discussed the importance of exposure as part of risk assessment. EFSA also outlined a plan to assess different human populations
-
Both EFSA and ICCVAM gave a detailed retrospective assessment of the historic and current use of NAMs
-
EFSA and the US EPA stated specific plans to implement the goals with the US EPA supplementing these with timelines for these plans
It should be noted that the need for an EU wide roadmap has been recognised in the New Approach Methodologies Workshop run by ECHA in 2023 and this may in part be achieved by one of the work packages of the PARC project.
5. Perspectives on specific NAMs
As previously stated the term NAM does not have a legal definition and can cover a range of different methodologies, including in silico, in chemico, in vitro methods, omics technologies, grouping and RxA strategies, as well as other emerging techniques and methods (ECHA, 2016c; US EPA, 2018; van der Zalm et al., 2022). In silico models utilise computer simulations and algorithm-driven predictions to assess toxicity, while in vitro methods employ cell-based assays to evaluate biological responses. In chemico techniques focus on chemical interactions, and omics technologies provide insights into biological pathways and mechanisms. Grouping and read-across strategies enable the extrapolation of data based on structural and functional similarities.
These methodologies are addressing several key limitations associated with traditional animal testing. Firstly, they offer alternative methods that reduce the number of animals used in toxicology studies, aligning with ethical concerns and regulatory pressures for humane scientific practices (Hilton et al., 2023). Secondly, NAMs can enhance the efficiency and accuracy of regulatory decision-making by providing faster and potentially more predictive data about human-relevant toxicological effects. However, despite their promise, NAMs require regulatory readiness and acceptance to be considered reliable and accurate methods.
This section comments on the current state of regulatory readiness and the associated expert discussion on the applicability of each type of NAMs. The key parameters specific to each NAM type which must be considered for validation and regulatory acceptance are discussed. Secondly, it gives an overview of how regulatory agencies evaluate the regulatory readiness of each NAM type including how each method is (or is proposed to be) validated and a qualitative description of the necessary steps required to enhance the regulatory readiness of NAMs.
Numerous tools have been developed to support the identification and use of NAMs. Many of these are collections of (Q)SAR and PBK models, but some tools to support the use of omics and identification of AOPs are also freely available. The models are often
linked to databases of toxicological results. A list of these tools and databases is included in Annex 4.
5.1. Regulatory Readiness Assessment of NAMs: a general discussion
The assessment of regulatory readiness of New Approach Methodologies (NAMs) is a crucial step towards their adoption and integration into regulatory frameworks. While NAM development has accelerated rapidly over the last several decades, validation, acceptance, and implementation of these approaches within the context of regulatory decision-making has not kept pace (Bal-Price et al., 2018; van der Zalm et al., 2022). Regulatory readiness is a term used across several industries and is described as the “stage of a process a technology is at and what are the steps needed to complete the regulatory steps to advance development of the product” (McGowran & Harris, 2020). There is no single approach to measuring regulatory readiness but in the context of this report, regulatory readiness is a measure of the progress through the validation protocols by which regulators gain confidence in a study. For each type of NAM addressed in this chapter, a consideration of the criteria identified by experts that would need to be met to increase regulatory readiness are discussed followed by a review of the current regulatory readiness that experts believe each NAM displays. Where appropriate, a more general discussion on the opportunities and barriers presented by each NAM is then included.
To gain regulatory acceptance and scientific confidence, NAMs must address five essential criteria outlined in van der Zalm et al.'s framework for establishing scientific confidence in NAMs (van der Zalm et al., 2022) (Table 5). These criteria, common to all types of NAMs, are based on roadmaps and evaluation frameworks from various organisations (Casati et al., 2018; ECHA, 2016b; EFSA, 2022; JRC, 2021a; OECD, 2021a, 2023a; US Consumer Product Safety Commission, 2022; US EPA, 2018; US FDA, 2017). They aim to advance the acceptance and use of NAMs by ensuring reliability and human relevance.
This means that the term regulatory readiness has a different meaning depending on the type of NAM and its proposed use (e.g. a NAM may be “ready” for use in screening but not for risk assessment). This should always be kept in mind when making this assessment and the context of use should be stated when a regulatory readiness is stated.
Escher et al. (2022) have published a very detailed assessment of the regulatory readiness of specific studies based on seven areas that are believed to require further scientific development for integration into feed and food risk assessments. They outline the process by which a study can be incorporated into regulatory frameworks (Figure 4).

This process can be applied to all NAM studies and approaches, but as discussed later in this report, it may be necessary for each stage to be done in a different way for different regulations or regulatory purposes. In their report Escher et al. use a traffic light system graduated based on the stages outlined in Figure 4 to describe the regulatory readiness of a method.
The literature review showed that assessments of the regulatory readiness of individual NAMs have been performed by several experts, it was decided that adding a duplicate assessment to these would not be useful. Instead we report the criteria that have been identified as been required in a NAM to make progress towards regulatory acceptance. The easiest level of regulatory readiness to demonstrate is where a method has been formally validated by an organisation such as the OECD (listed in Annex 6).
5.2. In Silico methods
In silico models in toxicology are defined as computational tools or approaches that utilise mathematical algorithms and computer simulations to predict toxicological outcomes or assess the potential hazards of chemical substances. These models are designed to mimic biological processes, molecular interactions, or toxicological pathways (Myatt et al., 2018). In silico models encompass a variety of techniques, including:
-
(Quantitative) Structure-Activity Relationship ((Q)SAR) models: (Q)SARs models predict chemical compounds’ biological activity or toxicity based on their molecular structure. (Q)SAR models establish quantitative relationships between the chemical structure of compounds and their observed biological effects, allowing for the prediction of potency, efficacy, and potential adverse effects. These in silico methods can be described as either a structure-activity relationship (SAR) model (qualitative prediction) or a QSAR model (quantitative prediction). The OECD and ECHA jointly manage the QSAR toolbox (OECD QSAR Toolbox, n.d.) which currently contains 254 models for the prediction of properties. (Q)SAR models can offer efficient screening of chemical libraries, guiding safer compound design and regulatory decision-making (Myatt et al., 2018; OECD, 2007, 2023c).
-
PBK models: These models, as defined in the OECD guidance document No. 331 on the characterisation, validation and reporting of PBK models for regulatory purposes, are computational frameworks employed to simulate the absorption, distribution, metabolism, and excretion (ADME) of chemicals within the body (OECD, 2021c). These models integrate anatomical, physiological, and biochemical parameters to predict the concentration-time profiles of substances in various tissues and organs. PBK models can serve regulatory purposes by aiding in dose optimisation, risk assessment, and decision-making regarding chemical safety and exposure (OECD, 2021c). These models can provide the link between cellular effects assessed by in vitro methods and the dose required to cause an adverse effect in an organism. Where these models are based on humans, they can provide a human-centric aspect of risk assessment that is hoped to mean NGRA is more relevant than the traditional animal based approach. It should be noted that other acronyms such as PBPK (Physiologically-based Pharmacokinetic), PBBK (Physiological Based Biokinetic) and PBTK (Physiologically Based Toxicokinetic) are used for these types of study but all will be grouped under the term PBK in this report, following the suggestion by Paini (2019).
-
Machine Learning Models: Some NAMs, such as omics and other high throughput screening, can generate a large body of data. When combined with the large number of independent studies using NAMs, the volume of data may be too high for individuals to transform it into meaningful information. In order to leverage large datasets to predict toxicological outcomes, machine learning models and artificial intelligence are being developed to facilitate their interrogation. These models are expected to be able to analyze chemical data, predict toxicity endpoints, and inform regulatory decision-making processes (Myatt et al., 2018).
The potential to use in silico methods is well established in existing regulatory frameworks, with the European Chemicals Agency producing a practical guide (ECHA, 2016d) for the use of in silico methods in both REACH and CLP . In this section, the regulatory readiness of in silico models, specifically (Q)SAR and PBK models, is evaluated.
5.2.1. Considerations for Regulatory Readiness Specific to In Silico Models
QSAR models
Validation and regulatory acceptance of (Q)SAR models necessitate consideration of several key parameters, in alignment with the OECD (Q)SAR Validation Principles outlined in OECD Guidance Document No. 69 (OECD, 2007) and OECD Guidance Document No. 386 (OECD, 2023c). These principles provide a robust framework for ensuring the reliability, transparency, and applicability of (Q)SAR models in regulatory contexts.
Principle 1 — Defined Endpoint
The first principle is to establish a defined endpoint, which specifies the biological activity or toxicological effect being predicted by the (Q)SAR model. This clarity ensures consistency and facilitates interpretation of the model’s predictions. The quality and relevance of input data are critical factors that influence the reliability and validity of (Q)SAR models. Therefore, careful selection, curation, and standardisation of structural descriptors and experimental endpoint measurements are necessary to ensure data quality, consistency, and representativeness, aligning with the OECD principle of a defined endpoint.
Principle 2 — Unambiguous Algorithm
An unambiguous algorithm is essential to ensure transparency and reproducibility in how the model transforms chemical structures into predictions. Transparency and documentation are fundamental aspects of (Q)SAR validation, necessitating detailed reporting of model development, validation procedures, and performance evaluation methodologies to facilitate reproducibility and expert review. Furthermore, it is also important to be transparent about the assumptions, limitations, and uncertainties associated with the (Q)SAR models. It is essential for stakeholders to make informed decisions regarding their regulatory acceptance and utilisation
Principle 3 — Defined Domain of Applicability
A defined domain of applicability is crucial to delineate the scope of chemicals and biological activities for which the (Q)SAR model’s predictions are valid. This principle emphasises the importance of considering both the structural diversity of chemicals and the biological mechanisms underlying the endpoint of interest. Additionally, to ensure that the predictions are reliable, the (Q)SAR model’s applicability domain should be thoroughly tested across different chemical structures and endpoints. To sum up, the applicability domain is defined based on empirical data and expert judgement and specifies the range of chemicals and biological activities for which the model’s predictions are valid.
Principle 4 — Appropriate Measures of Goodness-of-fit, Robustness, and Predictivity
Appropriate measures of goodness-of-fit, robustness, and predictivity are imperative for assessing the performance and reliability of (Q)SAR models. Statistical metrics such as accuracy, sensitivity, specificity, and concordance are commonly employed to evaluate the model’s predictive performance against experimental data.
Principle 5 — Defined Mechanism of Action
A mechanistic interpretation and defined mode of action provide essential insights into the underlying biological mechanisms through which chemicals exert their effects. Understanding these mechanisms enhances the scientific basis of (Q)SAR predictions and supports more informed risk assessments. Incorporating mechanistic insights into (Q)SAR model development and validation strengthens the reliability and regulatory acceptance of the models.
Furthermore, in addition to the principles mentioned earlier, continuous refinement and updates are crucial for the ongoing validation and regulatory acceptance of (Q)SAR models. It is important to regularly reassess and re-evaluate models in light of new data, scientific advancements, and evolving regulatory requirements to maintain (Q)SAR models relevance and reliability over time. This process of refinement and validation helps to boost the confidence of stakeholders and regulatory bodies in the predictive capabilities of (Q)SAR models (Myatt et al., 2018; OECD, 2007, 2023c).
PBK models
Validation and regulatory acceptance of PBK models involves two key parameters, as outlined in various guidance documents from national or supranational agencies. (EFSA, 2014; EMA, 2018; US EPA, 2006; US FDA, 2018; WHO, 2010). These parameters are also summarised in OECD Guidance Document No. 331 (OECD, 2021c).
Context & Implementation
The first parameter is to ensure that the PBK model is suitable for its intended purpose and context of use, it is crucial to define the scope of application, specify the regulatory questions being addressed, and consider factors such as data availability, computational resources, and user expertise.
Model Validity
Model validity involves assessing the scientific rigour and reliability of the PBK model. This includes accurately characterising the model structure and parameters to reflect physiological processes and capture interindividual variability. Validation should encompass a wide range of chemicals, exposure scenarios, and species to ensure applicability and reliability. Sensitivity analysis is essential to identify influential parameters and assess the model’s robustness to uncertainties.
Moreover, analogous to the validation of a (Q)SAR model (OECD, 2007) to demonstrate the scientific validity (reliability and relevance) of a PBK model, it is crucial to compare model predictions with observed data from relevant in vivo or in vitro studies and assess the model’s ability to reproduce (pharmaco)kinetic profiles under various conditions. Transparent reporting of model assumptions, limitations, and uncertainties further enhances the credibility and confidence in PBK model predictions.
Overall, adherence to these categories and considerations enhances the regulatory acceptance and utility of PBK models in informing chemical safety assessment decision-making.
5.2.2. Expert opinions of the Regulatory Readiness of In Silico Models
The literature review identified multiple publications and guidance documents that discuss the regulatory acceptance and readiness of in silico models. Annex 7 summarises some of the key publications and guidance documents identified.
The regulatory readiness of (Q)SAR and PBK models worldwide can be assessed based on key guidance documents and publications (see Table 2), particularly those from the Organisation for Economic Co-operation and Development (OECD) mentioned in section 1.2.1.2 above (OECD, 2007, 2021c, 2023c). The OECD plays an important role in developing internationally accepted guidelines for the validation and application of these modelling approaches in regulatory contexts.
QSAR models
The OECD’s Guidance document No. 69 on the validation of (Q)SAR models (2007) provides a robust framework for validating and applying (Q)SAR models in regulatory assessments. It emphasises the importance of predictive performance, reliability, transparency, and applicability of (Q)SAR models within regulatory contexts. While the OECD’s Guidance Document No.69 (OECD, 2007) focuses specifically on the validation of (Q)SAR models, it is important to distinguish its principles from those outlined in OECD Guidance Document No. 34 on the validation and international acceptance of new or updated test methods for hazard assessment (OECD, 2005a) Unlike OECD GD No.69, which concentrates on the validation of (Q)SAR models, OECD GD No.34 addresses the broader spectrum of test methods used in hazard assessment, encompassing experimental approaches. While both documents share common themes such as reliability, transparency, and applicability, OECD GD No.34 places additional emphasis on aspects specific to experimental methods, such as reproducibility, sensitivity, and specificity.
In Europe, ECHA places significant emphasis on (Q)SAR models for chemical hazard and risk assessment under the REACH regulation, aligning closely with the validation principles outlined in OECD Guidance document No.69 (OECD, 2007). ECHA has also issued guidance documents specifically for validating and accepting (Q)SAR models under the REACH regulation (ECHA, 2008, 2016b). For (Q)SAR predictions to gain acceptance under chemical frameworks such as REACH, biocides or cosmetics regulation, scientific validation is vital, with substances needing to fall within the applicability domain of the model (ECHA, 2008, 2016d; Gellatly & Sewell, 2019b) In addition, in the context of ECHA chemical regulations such as REACH or biocides, (Q)SAR predictions made by a scientifically validated (Q)SAR model are only accepted as standalone if they are capable of determining the classification and labelling of the substance and provide a conclusion on the hazard risk assessment. (Q)SAR predictions that are not suitable for classification and labelling or hazard risk assessment cannot be accepted alone and must be used as part of a WoE approach or as supporting information. (ECHA, 2008, p. 2016). In PPP regulations, (Q)SAR models are used for preliminary assessments but require additional data to fully replace complex in vivo tests (Benigni et al., 2019). To aid in the identification of validated (Q)SAR models, the EU Joint Research Centre (JRC) has developed a database (JRC, 2020), and information is structured using the (Q)SAR Model Reporting Format (QMRF) in accordance with OECD principles.
In the United States, regulatory agencies such as the Environmental Protection Agency (EPA) and the FDA increasingly integrate (Q)SAR predictions into chemical risk assessments, particularly under the TSCA reforms (US EPA, 2018) (Q)SAR methods offer a rapid and reasonably accurate means of prediction, contributing to regulatory decision-making across various contexts. This was confirmed in the stakeholder interviews discussed earlier.
Escher et al. (2022) do not make a comprehensive assessment of non-PBK models but do make the following important comments regarding the regulatory readiness of (Q)SAR for the use by EFSA
-
(Q)SAR models using data from the ToxCast/Tox21 database are available but their regulatory readiness ranges from research to regulatory acceptance. The models for estrogen and androgen receptors used in the risk assessment for endocrine disruptors have been fully adopted
-
Understanding regulatory relevance of a (Q)SAR is difficult when the data that is used to build the model is not freely accessible. The use of templates such as the QMRF is expected to help with regulatory acceptance. For other models such as REACH-across/RASAR they state that unless data curation is improved it is difficult to comment on the accuracy of any predictions
Although in silico methods are available for physicochemical, toxicological, ecotoxicological and environmental fate endpoints, and there is encouragement to use them as a replacement for in vivo studies, the requirements placed on (Q)SARs to allow them to be used as direct replacements may be too tight for them to be used extensively other than to address physicochemical endpoints. With EU REACH, ECHA gives extensive guidance on how to use (Q)SAR (ECHA, 2008) setting out to following stipulations for its use as a key study:
-
results are derived from a (Q)SAR model whose scientific validity has been established
-
the substance falls within the applicability domain of the (Q)SAR model,
-
results are adequate for the purpose of classification and labelling and/or risk assessment
-
adequate and reliable documentation of the applied method is provided
Within this guidance document, it is expected that few (Q)SAR tools will achieve all of these requirements but it does envisage that they would play an important role in a WoE approach or to supplement and support integrated testing strategies. Beyond registration, ECHA and the Member State Competent Authorities are known to use modelling software to inform substance evaluations that are used to identify whether additional regulatory management measures. For example, QSAR predictions for oestrogen receptor interference were used during the substance evaluation (ECHA, 2015) of 4,4’-propane-2,2-diyldiphenol, polymer with 2-methyloxirane to support the concern that the substance might display endocrine disrupting properties and to identify the grade of the substance that should be tested in further in vivo studies.
For other EU regulations REACH, in silico methods are especially important in regulations where animal studies are prohibited. (Gellatly & Sewell, 2019a) have reviewed the different types of in silico methods that can be used in meeting the obligations of the EU Cosmetics Regulation No. 1223/2009 (EC, 2009) but also discuss challenges for the cosmetics industry to apply these approaches. The note that although the Scientific Community on Consumer Safety (SCCS) Notes of Guidance (SCCS, 2006, 2018) acknowledge the importance of (Q)SAR, they are advised to be used alongside in chemico and in vitro studies as part of WoE. Outside the EU, the International Cooperation on Cosmetics Regulation(ICCR, 2014) report that in silico methods are largely being used by cosmetics companies for internal decisions rather than in regulatory submissions but it should be noted that this was published in 2014 and companies such as Unilever and L’Oreal are at the cutting edge of research into NAMs so this might be an outdated position.
Pesticides are applied to crops that are intended to be used as food ingredients and low levels of residues are likely even after processing. Although examining the toxicology of the active ingredient in a pesticide is a relatively straight-forward task, identifying and characterising the toxicology of its degradation products and metabolites is more difficult. This is because it is not a trivial matter to fully identify and isolate them in sufficient quantities required for in vivo studies. The use of (Q)SAR has been investigated to allow screening of a range of potential metabolites and degradation products to identify and prioritise those that present the greatest potential hazard to human health, especially those in vulnerable populations. Both Benigni et al. (2019) and Herrmann et al., (2020) have examined whether (Q)SAR tools used to predict mutagenic properties can be used for regulatory purposes. The studies concluded with similar suggestions.
-
Complementary (Q)SAR tools should be used instead of a single tool where possible
-
(Q)SAR tools have an advantage of other NAMs (e.g. RxA) in that their performance (sensitivity, accuracy, specificity) can be measured quantitatively and hence by validated
-
(Q)SAR can be used to predict in vitro bacterial mutagenicity with confidence but less so for other assays. From a regulatory context, the predictions for the Ames test could be used for prioritisation and as complementary information to other evidence
-
The quality of a (Q)SAR is dependent on the quality of the data used to build it. Data curation is of particular importance
-
As more data is generated and used in (Q)SAR tools, the greater the confidence there will be in their outputs if the quality of the data is maintained
-
Understanding the applicability domain of a (Q)SAR tool is essential, meaning this needs to be made explicit for any tools developed
-
Although the activity of using a (Q)SAR tool itself may not need as much expertise as other NAMs such as RxA, integrating their results with other evidence will still require expertise
In “Guidance on the establishment of the residue definition for dietary risk assessment” (EFSA PPR Panel, 2016), EFSA suggests the use of QSAR to support in establishing a TTC. The TTC approach is a pragmatic, scientifically valid methodology to assess the safety of substances of unknown toxicity found in food and the environment, recommended as a useful screening tool (EFSA, 2019). The application of the TTC concept utilises the classification scheme which was originally proposed by Cramer, Ford and Hall (Cramer et al., 1978) as a priority-setting tool and as a means of making expert judgements in food chemical risk assessment more transparent and reproducible. Czaja et al. (2020) recommends updating Cramer classification using recent advances possibly to account for particular populations (e.g. vulnerable groups such as infants and the elderly).
PBK models
The AOP approach takes a bottom-up approach to hazard and risk assessment by examining or modelling the effect of a chemical to increasingly complex biological entities along the pathway. Until the NAMs that address organs and the whole body become more advanced there currently needs to be an extrapolation to understand the impact on the whole organism. Most current regulations are intended to ensure a substance can be used safely, meaning that risk assessments are based on the exposure of the organism. The link between exposure of the organism and the exposure of the cell is provided by understanding of the toxicokinetics of the substance. Although there is a validated in vivo study to examine toxicokinetics (OECD TG 417) (OECD, 2010), the predictivity of animal studies for human exposure has been questioned (Musther et al., 2014). Under REACH there is a requirement to assess the toxicokinetic behaviour of the registered substance that applies to all substances registered at > 10 tonnes. The registrant has the discretion to decide how this assessment is conducted and can use expert judgement based on available data and modelling instead of animal studies. The regulators have not investigated the comparative quality of these approaches from a holistic perspective across a range of registration dossiers. A study into this could give a clearer idea of whether PBK models are being used in regulations and whether they are meeting the quality requirements outlined within this report. For NAMs to reduce or replace animal studies, the use of PBK models will be essential to estimate whole organism exposure from cellular exposure used in in vitro studies, referred to as IVIVE.
The OECD’s Guidance document on the characterisation, validation, and reporting of PBK models for regulatory purposes (2021b) provides comprehensive instructions for developing and validating PBK models. It emphasises characterising model uncertainty, documenting assumptions, and conducting sensitivity analyses to ensure robustness and reliability, facilitating their suitability for regulatory decision-making globally (OECD, 2021c). Additionally, the guidance document provides a list of eight case studies (OECD, 2021b).
PBK models are increasingly being developed and adopted across various sectors globally, particularly in Europe and the USA. Various guidance documents from national or supranational agencies are available (EFSA, 2014; EMA, 2018; US EPA, 2006; US FDA, 2018; WHO, 2010). However, the publications listed in Annex 7 outline the importance of validation and transparency of these models to be accepted by regulatory bodies.
On a more general side, contributions from experts in the field of in silico modelling, such as those outlined by Myatt et al. (2018), provide valuable insights for developing reliable in silico models. They emphasise the importance of standardised protocols for conducting toxicity-related predictions, covering aspects such as data curation, model development, performance evaluation, and regulatory applications. The standardised protocols also allow the evaluation of the level of confidence in the assessment based on the relevance and reliability of the information. These publications offer practical guidance for researchers and regulatory authorities to determine the reliability of in silico predictions alongside experimental data.
Unlike other (Q)SAR tools, Escher et al. (2022) devote an entire section to assessing the regulatory readiness of PBK models. They assess 33 PBK models, categorising them by route of exposure, organ and for which sector of use they are designed. The key message from this paper is that all PBK models are regarded as being either in a research or in situ validation phase. A key barrier to further progress is a lack of IVIVE approaches meaning reverse dosimetry cannot be used to estimate the dose at which an adverse effect may be observed. This is especially important for food and feed risk assessment where there is an absence of in vivo data against which to benchmark such an assessment.
Overall, while there is a clear trend towards increased regulatory acceptance of in silico models like (Q)SAR and PBK, particularly in countries with structured chemical regulatory frameworks, challenges such as model validation, defining applicability domains, and demonstrating relevance to human health persist. Further work is needed particularly for PBK models to be integrated into regulatory decision-making, a comprehensive evaluation framework addressing validity, contextual relevance, and confidence determination is essential for wider acceptance across all sectors and regions (OECD, 2021c; Paini et al., 2021). Paini et al. (2021) has proposed alternative approaches to validation that would reduce the need for new in vivo studies to provide benchmarks:
-
Uncertainty and sensitivity analysis
-
Use of MPS to parameterise the model
-
Use RxA to similar analogues with in vivo data
PBK models are widely used in the pharmaceutical sector during during the development of active pharmaceutical ingredients (APIs). These models include organ-specific simulations and are tailored for specific populations, such as pediatric patients (Deepika & Kumar, 2023). As these are used during the pre-market phase of development, it is unclear how many of these are validated to the extent required by regulations. Traditionally, PBK models are calibrated and evaluated against in vivo data in a test species, but this means their applicability for data-poor substances is less certain. As previously discussed, the applicability domain of a (Q)SAR model is fundamental to how it can be used so it is difficult to use models designed specifically for a different sector such as drugs or pesticides for cosmetic products. PBK models designed for other sectors are often based on oral exposure whereas the dermal route is most relevant to cosmetics. The fragrance ingredients in cosmetics can have a complex composition which many in silico methods are not designed to address. This has meant that in silico tools are more commonly used within a WoE or in internal decision-making context. Cronin et al., (2022) expands on the importance of PBK models to estimate realistic exposure to cosmetic ingredients and hence to calculate a TTC for cosmetic ingredients which can be used as a first-tier screen for the safety of such substances.
Despite the positive perception from the research community, a review by Punt et al. (2017) suggested that no (Q)SAR to predict kinetic data and very few PBK modelled prediction had been used in risk evaluations of pesticides, medicines or food, primarily being used to understand species differences or drug-drug interactions. To improve the use of this technology in regulatory risk assessment, guidelines and suggestions have been published on the reporting of modelling. Many of these are related to the pharmaceutical sector (EMA, 2018; US FDA, 2018) but the US EPA published a white paper on the use of PBK models to support derivation of Acute Exposure Guideline Levels (NCR, 2010).
5.3. Regulatory Readiness of In Vitro and In Chemico Test Methods
In vitro test methods are defined as experiments conducted outside of a living organism, using cells, tissues, or organs to study biological and toxicological effects under controlled conditions. These methods can play an important role in investigating biological pathways, and determining the safety of chemicals. Moreover, they serve as alternatives to traditional animal testing and are increasingly used to replace or reduce animal testing in regulatory contexts (ICCVAM, 2024; Petersen et al., 2023). The OECD emphasises that in vitro methods, when properly validated, provide crucial insights that can support regulatory decisions and scientific research (OECD, 2018a).
In chemico test methods are laboratory-based techniques that evaluate the interactions and reactivities of substances on a molecular or chemical level without using living organisms. These methods are primarily designed to provide mechanistic data that are essential for investigating and understanding chemical behaviour, including toxicity and transformation processes in biological systems. As a component of NAMs, in chemico testing can aid in understanding the mechanistic basis of chemical reactions essential for risk assessment and regulatory processes (ICCVAM, 2024; Petersen et al., 2023).
Both in vitro and in chemico test methods play pivotal roles in modern risk assessment frameworks and regulatory decision-making processes. These methods help to provide a better understanding of chemical behaviour and toxicity by uncovering the underlying mechanisms.
5.3.1. Considerations for Regulatory Readiness Specific to In Vitro and In Chemico Test Methods
To achieve validation and regulatory acceptance of in vitro test methods, certain critical parameters must be rigorously established, as highlighted in the OECD’s Guidance Document on Good In Vitro Method Practices (GIVIMP) (OECD, 2018a) and further exemplified by Bal-Price’s recommendations for in vitro test readiness in developmental neurotoxicity (Bal-Price et al., 2018). The OECD guidance document emphasises a comprehensive framework that prioritises reproducibility, reliability, and relevance, outlining the best scientific, technical and quality practices for developing and implementing in vitro methods that can be confidently used for regulatory and scientific purposes.
While the key parameters for achieving validation and regulatory acceptance of in vitro and in chemico test methods may differ due to differences in their methodologies and objectives, the validation process for both types of methods shares commonalities (OECD, 2005a). The reliability, relevance, and predictive ability of both types of methods are important in assessing chemical safety and toxicity. Therefore, in this section, the specific considerations for regulatory readiness of individual in vitro and in chemico test methods are based on the set of criteria outlined by Bal-Price et al., (2018), which are aligned with OECD Guidance on Guidance Document on Good In Vitro Method Practices (GIVIMP). These criteria are divided into three different phases to guide the development and implementation of the assays for regulatory purposes, as follows:
Phase I includes the basic key features of the test method as they would be provided by academic researchers such as:
-
Test system (only in vitro tests)
-
Exposure scheme (both in vitro and in chemico tests)
-
Documentation / Standard Operating Procedures (SOP) (both in vitro and in chemico tests)
-
Main endpoint(s) measured (both in vitro and in chemico tests)
-
Cytotoxicity (only in vitro tests)
-
Test method controls (both in vitro and in chemico tests)
-
Data evaluation (both in vitro and in chemico tests)
Phase II relates to the implementation of a test for practical applications in industry or for regulatory purposes and includes the following key criteria:
-
Detailed test strategy (both in vitro and in chemico tests)
-
Availability of test robustness data (both in vitro and in chemico tests)
-
Clearly defined test benchmarks (both in vitro and in chemico tests)
-
Detailed and available prediction model data (both in vitro and in chemico tests)
-
Clear and detailed description of the applicability domain (both in vitro and in chemico tests)
Phase III is related to the use of the assay for screening. This phase is optional as not each test method is used for a screening approach. It includes the following criteria:
- Clearly defined screening hits criteria and available data (both in vitro and in chemico tests)
Overall, adhering to these detailed parameters ensures reproducibility, reliability, and relevance of in vitro and in chemico test methods that can be validated to meet the high standards required for scientific and regulatory approval. This contributes significantly to advancing toxicological research and safety assessments while supporting the shift towards more ethical, animal-free testing paradigms.
5.3.2. Expert views on the regulatory readiness of in vitro and in chemico test methods
Several guidance documents and publications related to the validation, regulatory acceptance and readiness of in vitro and in chemico test methods have been identified. Annex 8 summarises some of the key publications identified. These guidance documents and publications provide insights into the regulatory acceptance and challenges associated with the integration of in vitro and in chemico test methods into chemical safety risk assessment. Moreover, ongoing developments in these areas are increasing through the development, validation, or validation processes of numerous test methods, as supported by scientific literature and regulatory guidelines. In Europe, the USA, Japan, and Korea, regulatory agencies responsible for validating alternative methods maintain lists of these methods, accessible through their respective websites:
-
Europe: EU Reference Laboratory for alternatives to animal testing (EURL ECVAM) provides the TSAR - Tracking System for Alternative methods towards Regulatory acceptance, accessible at https://tsar.jrc.ec.europa.eu/
-
USA: The NTP (National Toxicology Program) Interagency Center for the Evaluation of Alternative Toxicological Methods (NICETAM) provides information at https://ntp.niehs.nih.gov/whatwestudy/niceatm/accept-methods
-
Japan: The Japanese Center for the Validation of Alternative Methods (JaCVAM) provides information at https://www.jacvam.jp/en/list.html
-
Korea: The Korean Center for the Validation of Alternative Methods (KoCVAM) provides at http://116.67.77.115/kocvamen/article/list.do?boardKey=13&menuKey=24
The OECD’s Guidance documents (OECD, 2017a, 2018a) offers a framework for the validation and application of in vitro and in chemico tests in regulatory assessments. These documents outline principles for reliability, relevance, and predictive ability of both types of test methods in regulatory context globally (see section 1.2.3.1).
In Europe, regulations such as REACH explicitly recognise and accept alternative testing methods, including in vitro and in chemico assays, to meet data requirements for chemical registration (ECHA, 2016b). However, if an in vitro test suggests the absence of an intrinsic property, the standard in vivo test may still be required to confirm its absence, unless specific exceptions apply, such as when negative results are part of an integrated approach. ECVAM in the EU is responsible for the scientific validation of new alternative testing methods. This process involves test development, pre-validation, validation, independent assessment, and progression towards regulatory acceptance. Data generated from both validated and pre-validated in vitro methods can be used under REACH as standalone, provided that the assay is adequate for classification, labeling, and risk assessment purposes and sufficient documentation for independent evaluation. Non-pre-validated data can only serve as supportive information, such as within a WoE justification (ECHA, 2016a). In the US, as outlined on the NICETAM webpage (NICEATM, n.d.), acceptance of in vitro and in chemico test varies across regulatory agencies, and initiatives like ToxCast and Tox21 have demonstrated the feasibility and utility of high-throughput screening assays. However, their integration into decision-making is still ongoing. In other regions such as Asia acceptance varies, with some countries making progress while others are exploring applicability (JaCVAM, n.d.-a, n.d.-b; KoCVAM, n.d.).
Moreover, the regulatory readiness for in vitro and in chemico tests varies depending on the specific chemical framework and the endpoint being evaluated. For instance, in vitro tests assessing skin corrosion or irritation typically encounter higher acceptance levels compared to those assessing more complex endpoint such as systemic toxicity or carcinogenicity. As demonstrated by Bal-Price et al. (2018) for complex endpoints like DNT, standalone in chemico or in vitro tests are insufficient and must be used in combination with others, requiring a rigorous validation process for each test in the series. This process, described in OECD Guidance document No.34 on the validation and international acceptance of new and updated test methods for hazard assessment can take several years (OECD, 2005a). To accelerate this process Bal-Price et al. (2018)) recommend developing a framework of criteria for in vitro tests readiness highlighting the need to update the classical validation approach in view of innovative approaches that employ testing strategies involving combinations of tests rather than a single individual assay replacing an animal study (Bal-Price et al., 2018). Ongoing efforts aim to develop and validate alternative methods tailored to address complex endpoints as well as updating the classical validation method, with the goal of enhancing their regulatory applicability and acceptance across various chemical frameworks and accelerating the traditional validation process (Crouzet et al., 2023; Krebs et al., 2019; Mathisen et al., 2023).
Overall, there has been notable progress in promoting the use of in vitro and in chemico tests in chemical risk assessment. However, to ensure their use for more complex endpoints, widespread adoption and acceptance globally, across different regions and chemical frameworks, further collaboration, standardisation, and regulatory update and harmonisation efforts are required. The range and variety of in vitro and in chemico studies means that it is not possible to present a general discussion on their use in regulations beyond those already described in the section.
5.4. Regulatory Readiness of omics technologies
Omics technologies are advanced scientific methods that analyse the complex layers of biological molecules in living organisms. These methods include “the four big omics”: genomics, transcriptomics, proteomics, and metabolomics which analyse DNA, RNA, proteins, and metabolites, respectively, providing a holistic view of biological systems (Dai & Shen, 2022) They also include epiomics, such as epigenomics, epitranscriptomics and epiproteomics, and the category of interactomics, such as DNA-RNA interactomics, RNA-RNA interactomics, DNA-protein interactomics, RNA-protein interactomics, protein-protein interactomics and protein-metabolite interactomics (Dai & Shen, 2022). Then there are the so-called “knowledge-based omics”, which are developed to understand a specific field of knowledge in a systematic way, such as immunomics, microbiomics, exposomics, and many others (Dai & Shen, 2022). They can all help scientists understand the underlying mechanisms of toxicology and toxicity pathways, which are necessary for more accurate risk assessment (CATTPTRA-NRC, 2007; OECD, 2005b; Sauer et al., 2017). In addition, the term ‘multiomics’ used in the review article by Canzler et al. (2020) on multi-omics in toxicology, refers to the integration of two or more of these omics methods, enabling the simultaneous analysis of several layers of molecular information to provide a holistic understanding of complex biological phenomena.
Furthermore, as detailed by Harrill et al. (2021), omics technologies have become crucial in regulatory toxicology for mapping out the intricate molecular pathways and responses triggered by various substances (Harrill et al., 2021).
5.4.1. Considerations for Regulatory Readiness Specific to Omics Technologies
ECETOC held a workshop in October 2016 to discuss the regulatory acceptance of omics data. The discussions revealed that best practices should be followed when performing omics studies for regulatory purposes. However, it was concluded that overly prescriptive guidance for collecting and analysing omics data might not be helpful, as omics technologies are constantly evolving and fit-for-purpose approaches are often required. Instead, a reporting framework was suggested as a way forward to describe how omics data are generated, processed, analysed, and stored (Buesen et al., 2017). This led to the development of a reporting framework and a guidance document by OECD (2023b) to facilitate regulatory acceptance of omics. The framework consists of four reporting modules that outline key parameters to be considered for regulatory acceptance of omics as follows:
-
The Study Summary Reporting Module (SSRM) provides a high-level overview of the regulatory toxicology and Omics experiment
-
The Toxicology Experiment Reporting Module (TERM) reports the key descriptors of the in vivo or in vitro toxicology study
-
Data Acquisition and Processing Reporting Modules (DAPRM) report descriptions of the Omics assays, data acquisition and associated data
-
Data Analysis Reporting Modules (DARM) describe the statistical analysis that has been undertaken in the omics study
The OECD Guidance document No. 390 (OECD, 2023b) on reporting elements for the regulatory use of omics data from laboratory-based toxicology studies describe each of the reporting modules in detail.
5.4.2. Expert views on the regulatory readiness of omics technologies
The literature review identified multiple publications that discuss the regulatory acceptance and readiness of omics technologies. Annex 9 below summarises some of the key publications identified
These publications provide insights into the regulatory acceptance and challenges associated with the integration of grouping chemicals and read-across into chemical safety risk assessment.
The regulatory readiness of omics technologies is outlined primarily in OECD Guidance document No. 390, which acknowledges their increasing application in both research and regulatory toxicology for understanding toxicity mechanisms (OECD, 2023b). While mature omics technologies are widely used in research, their integration into regulatory decision-making remains limited (OECD, 2023b). To achieve validation and acceptance for regulatory use, Omics technologies must meet key parameters outlined in OECD Guidance No.390 and listed in the section 1.2.3.1 of this report. These technologies must also adhere to Good Laboratory Practices (GLP) to ensure that Omics studies are conducted under rigorously controlled conditions to guarantee the quality, reliability, and consistency of the data (Kauffmann et al., 2017). However, there are two significant barriers that hinder the routine application of Omics in regulatory decision making (OECD, 2023b). These two key challenges are:
-
The lack of transparency for data processing methods used to convert raw data into an interpretable list of observations; and
-
The lack of standardisation in reporting which makes it difficult for stakeholders, including regulators, to review the omics data, associated metadata, and the methodologies used to generate results
Moreover, Fortin et al. (2023) emphasise the significance of having a well-defined applicability domain, which is not easy to define explicitly for Omics due to the complexity and variability of biological systems. Although there are established criteria for data quality and reproducibility, they can significantly vary between different types of omics analyses, and there is a lack of standardisation across various regulatory bodies (Fortin et al., 2023) These parameters, such as clearly defined applicability domain and data acceptance criteria, are crucial for integrating technologies into regulatory frameworks. They ensure that the generated data are scientifically valid and reliable for risk assessment purposes. Escher et al. (2022) do not measure the regulatory readiness but do comment that despite their widespread use in research, omics are rarely used in regulatory decision-making. The development of reporting standards will be an important step towards this evolution.
Overall, publications listed in Annex 9 indicate that omics are increasingly being developed and adopted due to their excellent ability to predict toxicity. Their ability to measure molecular initiating events and downstream molecular phenotypes that predict toxicity is useful for identifying hazards (or adverse outcomes), analysing the mode of action, grouping chemicals for RxA and characterising potency through omic points-of-departure. However, more work is needed to integrate Omics into regulatory decision-making. There is a notable need for improvement in defining clearer applicability domains, data acceptance criteria, and standardisation in reporting and result interpretations. Further standardisation and guidance documentation may be necessary to fully integrate omics technologies into regulatory decision-making frameworks across different chemical application domains and regions.
Omics is a term that is used to describe several different techniques. One aspect that will need to be addressed to allow integration into regulations is to decide whether one particular technique is used in isolation or whether a multiomics approach to link two or more methods to give a holistic view of the biological interactions and consequences arising from exposure to a substance at a cellular level. A regulatory stakeholder in the interviews suggested that the multiomics approach was most likely to succeed with some techniques (e.g. proteomics and transcriptomics) needing to be used in conjunction. This perspective is reinforced in the conference report by Wojewodzic & Andreassen (2022) where one speaker “emphasised the importance of using the powerful combination of expressed genes and metabolic biomarkers (multi-omics approach), to reduce uncertainty and increase confidence in the prediction of an adverse outcome.” Elsewhere in this workshop report omics were described as “a powerful tool for grouping, read-across, and for obtaining a mechanistic understanding of a chemical’s MoAs.”. However a representative from ECHA said that omics data were rarely used in dossiers possibly due to a lack of confidence from registrants or evaluators and a lack of transparency regarding data gaps. Solutions to addressing datagaps in a multiomics approach have been suggested by Henao et al., (2023) but the regulatory acceptability of their approach has not been tested. In addition the links from adversities seen in each study to both each other and to any apical events in the whole organisms need to be fully explained and that the relevance of the responses to a stressor seen in the omics need to be shown to be relevant to the toxicological space under examination. It was noted that omics could be used to identify “non-toxicity” which can be difficult for NAMs to conclusively demonstrate, but this would need good benchmarking against known non-toxic substances. This perspective is reinforced by Gant et al., (2017).
5.5. Regulatory Readiness of Grouping and Read-Across (RxA)
The NAMs discussed until now are stand alone methods that can be used as part of a battery of tests to directly estimate the toxicological properties of a substance. Grouping and RxA can instead be regarded as a strategy to justify the use of existing data or to reduce the number of substances tested, as it uses knowledge of the basic biological and physicochemical parameters of more than one substance to be able to demonstrate similarity between the substances. It allows for the prediction of hazard information for one chemical (target chemical) based on data from analogous chemicals (source chemicals) within the same group or category based on the principles that substances with structural similarity are likely to exhibit similar (eco)-toxicological properties. There are two main types of grouping approaches used to predict the hazards of chemicals: the analogue approach and the category approach (ECHA, 2017; OECD, 2017d).
-
Analogue Approach: Data from one or more similar source chemical(s) are extrapolated to predict the hazard of a target chemical. This is done based on the assumption that the chemicals share (eco)-toxicological properties due to structural similarity
-
Category Approach: Chemicals are grouped based on similarities. Data from representative chemicals within the category are then used to predict the hazard of others chemicals within the same group based on the assumption they share (eco)-toxicological properties within the group
It is important to note that structural similarity is a pre-requisite for any grouping and RxA approaches (ECHA, 2017). The similarities may be due to a number of factors such as:
-
Common functional group (i.e. chemical similarity within the group)
-
Common precursors and/or likelihood of common breakdown products through physical and/or biological processes which result in structurally-similar degradation products (i.e. similarity through (bio)transformation)
-
A constant pattern in the changing of the potency of the properties across the group (i.e. of physicochemical and/or biological properties)
Any type of information can be used to justify grouping, so although NAMs can play an essential part of the process, existing animal study data can also be used. The ability to use grouping and RxA is explicitly stated in many regulations. For example, Annex XI of EU REACH states the situations where RxA can be used to fulfil a registration endpoint, stating “Substances whose physicochemical, toxicological and ecotoxicological properties are likely to be similar or follow a regular pattern as a result of structural similarity, may be considered as a group, or category, of substances. Application of the group concept requires that physicochemical properties, human health effects and environmental effects or environmental fate may be predicted from data for reference substance(s) within the group by interpolation to other substances in the group (read-across approach). This avoids the need to test every substance for every endpoint.” RxA should be made at an individual endpoint perspective although data used to justify the approach can be re-used across multiple RxAs if relevant.
5.5.1. Considerations for Regulatory Readiness Specific to Grouping and RxA
To facilitate regulatory acceptance, a RxA prediction needs to be justified in all aspects. Briefly, the justification of a RxA prediction needs to be robust, reliable and easily explained. The key parameters for regulatory readiness of grouping chemicals and RxA described below are based on the ECHA recommendations to registrants on performing and documenting a good-quality RxA (ECHA, 2013) and outlined in Schultz et al., (2015) workflow for reporting a RxA prediction in the regulatory context.
According to Annex XI, section 1.5 of the REACH regulation, a RxA approach must:
-
Be adequate for the purpose of classification and labelling and/or risk assessment
-
Have adequate and reliable coverage of the key parameters addressed in the corresponding test method referred to the hazard being predicted
-
Cover an exposure duration comparable or longer than the corresponding test method referred to the hazard being predicted
-
Adequate and reliable documentation of the applied method shall be provided
To document a RxA prediction, the following elements are considered essential:
-
A clear RxA hypothesis
-
A clear justification for the RxA hypothesis, based on available experimental data, especially for the source substance(s), including detailed key principles of structural similarity, reactivity, toxicokinetics, and metabolism of analogue(s)/category members, as well as similarity or trend in the physical-chemical properties
-
A list of all the substances (target(s) and source substance(s)) included in the approach
-
A detailed substance identity information of all substances included in the approach
-
An outline and assessment of the structural similarity(ies) between the substances
-
A list of the endpoints that are to be RxA
-
A data matrix for assessing similarity
-
A conclusion on the applicability of the proposed RxA approach
The use of grouping chemicals or RxA approaches in the regulatory context also requires consideration of the following additional parameters (ECHA, 2013, 2016d, 2017; OECD, 2017d; Schultz et al., 2015).
-
Applicability domain: It is specific to the category approach. In a category approach, the applicability domain is described by inclusion and/or exclusion rules that identify the extent of values for category members within which reliable predictions can be made
For example, the range of 1-octanol/water partition coefficient values, functional groups or carbon chain lengths within which the category is appropriate
-
Purity/impurities: For each analogue or category member, a purity/impurity profile needs to be catalogued. The potential impact of impurities on the endpoint(s) being considered in the RxA prediction should be identified
-
Uncertainty: Sources of uncertainty need to be identified and accommodated; these can typically be divided into two main types: (1) the uncertainty associated with the justification of similarity between the source and target structures, and (2) the uncertainty associated with the application of the particular RxA exercise
5.5.2. Expert views on the regulatory readiness of grouping chemicals and RxA
Guidance documents and publications related to the regulatory acceptance, readiness, and application of grouping chemicals and RxA have been identified. Annex 10 summarises some of the key publications identified.
As grouping and RxA is a methodological approach rather than a single study, it will vary in every case, so the term validation in the context of ensuring standardisation does not apply completely. Instead there is a drive to produce standard protocols and criteria to ensure that RxA arguments are well justified from a scientific point of view. RxA is a well-established technique for filling data gaps in hazard identifications and characterisation assessments for regulatory purposes, with varying degrees of acceptance and utilisation across different regions. Worldwide, OECD provides a harmonised guidance on grouping chemicals that can be used by OECD member countries (OECD, 2017d). In Europe, ECHA’s Read-Across Assessment framework (RAAF) is widely recognised and implemented within REACH regulation (ECHA, 2017). The RAAF was designed to enable evaluators to assess RxA arguments in REACH and BPR dossiers, but is expected to be used by registrants to optimise their use of the approach. The framework identifies assessment entities (AE) that need to be addressed in the justification for a RxA. The exact AE that should be used depend on the type of RxA and for which endpoint the RxA is being used, for example:
-
Identity and characterisation of the source substance
-
Link of structural similarities and differences with the proposed prediction.
-
Reliability and adequacy of the source study
-
Bias that influences the prediction
Despite this support, research by (Macmillan et al., 2024) has shown that the use of RxA to fulfil endpoints in registration dossiers has fallen from 27.7 % to 22.8 % between 2016 and 2022, indicating a reluctance (or a perception by registrants) on the part of the agency to accept RxA. They also cite an example of a compliance check of benzoic acid, C15-15 alkyl esters (CAS 68411-27-8) whereby the use of RxA to satisfy multiple endpoints was challenged by ECHA and an OECD TG 210 fish, early life stage (FELS) toxicity test was required. Although an improved RxA hypothesis was proposed, most registrants decided not to use this approach and proceeded with the study, with fear of rejection and enforcement action being the key reason. The studies confirmed the position of the improved RxA hypothesis meaning that ~150 vertebrates may have been used for no tangible benefit.
RxA is also used in other EU regulations. The meeting report on “Regulatory Acceptance of Read-Across” (Chesnut et al., 2018) hosted as part of the 56th Annual Meeting of the Society of Toxicology describes how EFSA have used RxA to analyse flavouring substances by placing over 2,600 substances into 34 chemical groups for assessment. One group, consisting of α,β-unsaturated carbonyls, was predicted to be genotoxic by RxA and the TTC for group members determined accordingly.
The formal integration of RxA into regulatory dossiers in the US is primarily utilised within specific EPA programs like the Pre-Manufacture Notice (PMN) process and the Toxic Substances Control Act (TSCA), with a notable absence of formal guidance documents (Patlewicz et al., 2019; Rovida et al., 2020) Patlewicz et al. gave an extensive review of how RxA is used by different US federal agencies, including Agency for Toxic Substances and Disease Registry (ATSDR), Department of Defense (DoD), EPA Office of Pollution Prevention and Toxics (EPA OPPT), National Center for Environmental Assessment (NCEA) and National Library of Medicine (NLM). Whilst most agencies used RxA for screening, prioritisation and to support emergency response, an risk assessment of n-heptanal by the EPA used RxA to other short-chain aldehydes, in part by demonstrating similar metabolic rates of n-heptanal with the source substances to detoxify the substance. Within ICCVAM, a Read-Across Workgroup (RAWG) has been established to examine the potential for use of RxA and areas where improvements are needed across US federal agencies. The RAWG identified the following concerns around the use of RxA:
-
The robustness of prediction by some tools is not easily understood
-
Understanding of the uncertainty in RxA is not clear enough to be used for some regulatory decisions
-
Although use of RxA to assess mixtures would be a great use, there was minimal guidance on how to do this
-
Quality of the source substance’s in vivo data can be inconsistent. Improvement in the detail and scope of such data would increase confidence in RxA
In other regulatory regimes, Canada has been progressively incorporating RxA into risk assessment programs, particularly through initiatives like the Chemicals Management Plan (CMP) (Rovida et al., 2020). Similarly, regulatory bodies in Asia, including those in China, Japan, and Korea, are increasingly adopting RA methods supported by national guidelines and international collaboration efforts (Gao et al., n.d.; Lee et al., 2022; Rovida et al., 2020). Lee et al. reviewed the use of RxA in South Korea in 2022, stating that “the Ministry of Environment (MOE) established the Act on Registration and Evaluation of Chemicals (AREC) in 2019 to enable registrants to submit alternative data such as information from read-across instead of in vivo data to support hazard assessment and determine chemical-specific risks. Further, the Ministry of Food and Drug Safety (MFDS) began to consider read-across approaches for establishing acceptable intake limits of impurities occurring during pharmaceutical manufacturing processes under the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) M7 guideline.” However, they also report that a lack of standardised acceptance criteria, inconsistencies in evidence robustness and poor reliability of the source data means that acceptance is likely to vary between different agencies.
In other regulatory regimes, Canada has been progressively incorporating RxA into risk assessment programs, particularly through initiatives like the Chemicals Management Plan (CMP) (Rovida et al., 2020) Similarly, regulatory bodies in Asia, including those in China, Japan, and Korea, are increasingly adopting RA methods supported by national guidelines and international collaboration efforts (Gao et al., 2020; Lee et al., 2022; Rovida et al., 2020). Lee et al. reviewed the use of RxA in South Korea in 2022, stating that “the Ministry of Environment (MOE) established the Act on Registration and Evaluation of Chemicals (AREC) in 2019 to enable registrants to submit alternative data such as information from read-across instead of in vivo data to support hazard assessment and determine chemical-specific risks. Further, the Ministry of Food and Drug Safety (MFDS) began to consider read-across approaches for establishing acceptable intake limits of impurities occurring during pharmaceutical manufacturing processes under the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) M7 guideline.”. However, they also report that a lack of standardised acceptance criteria, inconsistencies in evidence robustness and poor reliability of the source data means that acceptance is likely to vary between different agencies.
Regulatory acceptance and scientific confidence in RxA approaches rely on the clear reporting of defined parameters (see section 1.2.4.1 of the report) established in comprehensive regulatory guidelines and publicly available literature. The foundation of grouping chemicals in RxA is based on chemical similarity, in structure, physicochemical properties, functional groups, and metabolic pathways, requiring a robust rationale (OECD, 2017d). This similarity is pivotal in supporting the hypothesis that the grouped substances possess comparable toxicological and environmental behaviours. The RAAF (2017), Bergreen et al., (2015) and Schultz et al. (2015) emphasise the importance of comprehensive and reliable data for the source chemicals, justified analogue selection, and explicit uncertainty analysis to enhance the credibility of RxA conclusions, facilitate the regulatory review process and building trust in the RxA outcomes (Berggren et al., 2015; ECHA, 2017; Schultz et al., 2015).
Although Escher et al. (2022) regard the regulatory readiness of RxA as “in situ validation”, they make a very comprehensive assessment of the unpinning AOPs and of the approaches that can be used to build a RxA such as IATA, DA and tiered approaches. They assess over 10 AOPs as having regulatory acceptance or in independent review for single organ repeat exposure, endocrine disruption and DNT, which others at these stages for non-genotoxic carcinogenicity, development and reproductive toxicity and immunotoxicity.
The perceived reluctance of regulatory agencies to accept RxA in lieu of animal studies on a substance has driven several research projects to examine ways to develop and improve protocols to justify this approach to risk assessment. Perhaps the most comprehensive discussion was made by (N. Ball et al., 2016) in their paper entitled “Towards Good Read-Across Practice (GRAP)”. In this paper they make the following suggestions:
-
Comprehensive substance identification of both source and target substances is fundamental to successful RxA. This is particularly important for complex substances such as UVCBs (unknown or variable composition, complex reaction products or of biological materials). The poor quality of test substance characterisation in REACH dossiers was highlighted in the ECHA report on the role of robust study summaries (RSSs) in hazard assessment (ECHA, 2022)
-
Use ‘big data’ from high throughput screening and omics to establish biological similarity
-
Use in vitro studies to demonstrate local validity
-
Understand and report uncertainty around the RxA. Validation and standardisation of an individual study allows a user to understand the type and scale of uncertainty in their results. As RxA uses multiple studies, expert opinion and are often bespoke to the substance in question, these uncertainties are less easy to quantify. Uncertainty is not a well-defined concept but (Escher et al., 2019). present a description, using “referring to all types of limitations in available knowledge that affect the range and probability of possible answers to an assessment question” and clarify that it covers both the source of uncertainty and its impact on any conclusions from the assessment
-
Build a library of fit-for-purpose tools to support RxA
-
Ensure consistent approach to reporting and assessing RxA
The SEAURAT-1 projects, funded by the European Commission and Cosmetics Europe designed a series of case studies to examine the use of RxA in different scenarios (Berggren et al., 2015). The results of some of these case studies that addressed 90-day rat repeated dose toxicity demonstrated that toxicokinetic and/or toxicodynamic similarity is an essential component of a RxA justification and, while NAMs such as high throughput screening, in vitro and in silico methods can build the toxicodynamic rationale, toxicokinetic similarity will influence the degree of uncertainty over the uncertainty in a RxA conclusion (Schultz & Cronin, 2017). Another EC funded project, EU-ToxRisk, focused on developing a science-based strategy for RxA that incorporated NAMs. In a report of the project’s findings, Moné et al., (2020) reported on both the research done during the project and views from different stakeholders. It was suggested that the use of NAMs could reduce uncertainties inherent in the ‘classical’ approach to RxA by aligning with the AOP approach, namely:
-
Address metabolism of the source and target substances
-
Improve estimation of relative potency of the source and target substances by examining their relative potency at KE or MIE
-
Identify alternative mechanisms or targets
-
Potential to anchor results from a human context
In order to achieve these gains, especially from a regulatory context the following recommendations were made:
-
Sharing of data, test methods, results and knowledge is vital. This should be facilitated by using harmonised templates to report data and methods
-
Strengths and weaknesses, including uncertainty reporting, must be included alongside methodology. This would be particularly important where methods have not been validated. Escher et al. (2022) have suggested a structured and transparent method to report uncertainty for both RxA approaches and an AOP in general, but its acceptance within regulatory bodies is not clear
-
Inclusion of regulators in the design and application of AOPs from an early stage would improve confidence. Macmillan et al. (2024) build upon this by suggesting that communication should be two-directional and regulators should provide more clarity in what they expect in a RxA and publish examples when RxA has been accepted in a regulatory submission. Sewell et al. recommended establishing ‘safe havens’, where the results of parallel assessments could be discussed between researchers and regulators without an impact on any regulatory decisions
-
The AOP approach sets a structure for the RxA, however the amount of testing should be limited to that sufficient to give confidence in the RxA justification
Even should these recommendations be put in place, the authors identified areas where there is still research required:
-
NAMs are required to relate more closely to human toxicology instead of mammalian toxicology
-
Deriving a mechanistic hypothesis for an AOP is not simple for compounds with little or no prior information
-
A strategy to use NAMs and RxA to define substances with low or no toxicity needs to be developed
Grouping and read-across frameworks have been developed for specific classes of substance. The GRACIOUS Framework (2021) presents a way to write and test hypotheses for the grouping of different nanoforms, both for regulatory and Safe-by-design purposes. A user develops their own or uses pre-existing IATA to take a tiered approach to testing to allow the grouping of different nanoforms. Although it does not rule out the commissioning of new animal tests, it is intended to significantly reduce the need for them and to make full use of existing data.
5.6. Regulatory Readiness of Other NAMs
The field of toxicology is constantly evolving, and new approaches are being developed to transform chemical safety assessments. Some of these new methodologies include machine learning (ML), artificial intelligence (AI), and organ-on-a-chip (OoC) technologies. They are being increasingly recommended to be integrated into regulatory frameworks, reflecting a shift towards more innovative, efficient, and ethical approaches.
Machine learning and artificial intelligence are computational models that use algorithms to predict the effects of chemicals based on data-driven insights. These technologies can analyse vast datasets to identify patterns and predictions that are not immediately apparent to human researchers. Machine learning is a subarea of artificial intelligence, and it refers to mathematical or computer algorithms designed to teach or train a computational model to solve a problem or perform complex tasks based on some input parameters (Lin & Chou, 2022). Organ-on-a-chip, on the other hand, is a microfluidic device that simulates human organ functions on a chip, providing a dynamic model to study the systemic effects of substances in an integrated and human-relevant context (JRC, 2021b).
5.6.1. Considerations for Regulatory Readiness Specific to Other NAMs
For NAMs to be validated and accepted by regulatory authorities, they must meet several key parameters as described at the top of section 1.2. These include specifically demonstrating reproducibility, reliability, and predictive accuracy. The models must be transparent and interpretable, especially for AI, where the decision-making process should be understandable to ensure trust and acceptance. For organ-on-a-chip, the technology must replicate human physiological responses accurately enough to be considered a valid model for human health risk assessment. Validation involves comparing results with known clinical or toxicological outcomes to establish reliability (Holzer, 2023).
5.6.2. Expert views on the regulatory readiness for other NAMs
The literature review identified several publications that discuss the regulatory acceptance, readiness, and potential application in risk assessment of emerging technologies such as artificial intelligence (AI), machine learning (ML), and organ on a chip (OoC). Annex 11 summarises some of the key publications identified.
These publications provide insights into the regulatory considerations, challenges, and opportunities associated with the integration of machine learning organ-on-a-chip and AI, into chemical safety risk assessment.
Organ-on-a-chip
OoC technologies have great potential in various scientific domains and could reduce reliance on animal testing by replicating organ dynamics and physiological responses in a controlled microenvironment. The regulatory readiness of OoC is discussed by the JRC in a report summarising a workshop on standardisation of OoC (JRC, 2021b). In this report, it is concluded that despite their promises and widespread development, the regulatory readiness and adoption of OoC technologies face significant challenges.
Challenges:
-
Complexity and Cost: End-users (researchers, scientists, and professionals involved in safety evaluation) express reluctance due to the complexity, uncertainty in outputs, and high costs associated with OoC models
-
Developer-Centric Approach: The current development and design of OoC technologies are primarily driven by the developers, lacking collaboration with end-users to address practical needs, challenges, or preferences of end-users
-
Standardisation: Lack of standardised frameworks hinders widespread adoption, requiring thorough characterisation of OoC components across different technology readiness levels (TRLs)
Addressing these challenges requires concerted efforts and collaborative approaches. Initiatives like the European Organ-on-Chip Society (EUROoCS) and government-sponsored programs such as the NCATS in the United States aim to enhance development, testing, and implementation of OoC models. Public-private partnerships, like the Tissue Chip Validation Centre, facilitate collaborative testing and validation, promoting industry and regulatory acceptance (JRC, 2021b). Furthermore, participants of the workshop emphasise the importance of translating scientific evidence into regulatory standards to drive acceptance and foster a robust marketplace for human-relevant alternatives to animal testing. Moving forward, a strategic roadmap is needed to prioritise standardisation needs and align them with the various chemical frameworks strategies and related policies, ultimately realising the potential of OoC technology to transform risk assessment.
Escher et al. (2022) describes the state of the art biological systems to understand toxicodynamics for organs including the microbiome, intestinal tract, liver, kidney and heart from the perspective of testing food safety. These include 2-D, microporous filters, 3-D scaffolds and organ-on-a-chip cell arrays. They comment that these systems are not yet ready for toxicological screening and need to be part of a tiered testing in the near future. The need for a common genetic stock for all the different cells is highlighted.
Machine Learning and Artificial Intelligence
ML and AI are used for predictive modelling and data mining in toxicological assessments. In the context of toxicological assessments, this means that these technologies are used to analyse data related to the safety and potential toxicity of chemical substances by analysing large amounts of data, ML and AI can help identify potential hazards, predict toxicological outcomes, and support decision making related to chemical safety. Several publications on the applications of ML in various chemical frameworks were identified (Annex 11). However, no assessment of the regulatory readiness of these technologies or regulatory guidelines were found in the public domain. For artificial intelligence, a lot of data retrieving the use of AI as a tool in diverse domains such as scientific literature review were identified (Blümmel et al., 2023, 2024; Zgheib et al., 2021). Participants of a JRC workshop on artificial intelligence for Chemical Risk Assessment proposed a framework to adopt AI in chemical risk assessment (JRC, 2019). The framework includes the following criteria to use AI in Chemical Safety Risk Assessment:
-
Data Availability and Quality: AI can be employed to analyse large datasets for chemical safety risk assessment, provided that high-quality data is available. This includes data on chemical properties, toxicity, exposure levels, and environmental impact
-
Interpretability and Explainability: AI algorithms can be developed to provide interpretable and explainable results in chemical safety risk assessment. This involves designing models that offer insights into how predictions are made, allowing for transparency and understanding of the decision-making process
-
Regulatory Compliance: AI technologies can assist in ensuring regulatory compliance by automating processes related to data management, risk assessment, and reporting. This includes aligning AI-driven assessments with regulatory guidelines such as REACH and TSCA
-
Uncertainty and Confidence Estimation: AI models can be enhanced to quantify uncertainty and provide confidence estimates for their predictions. This involves implementing techniques for uncertainty quantification, such as probabilistic modelling and sensitivity analysis, to assess the reliability of model outputs
-
Generalisation and Transferability: AI algorithms can be designed to generalise across diverse chemical compounds and contexts. This includes developing models that can transfer knowledge and insights learned from one dataset to another, improving their applicability and utility in different scenarios
-
Ethical and Societal Implications: AI technologies must address ethical and societal concerns related to data privacy, algorithmic bias, and transparency. This involves integrating principles of fairness, accountability, and transparency into the design and deployment of AI systems to ensure responsible and ethical use in chemical safety risk assessment
These parameters outline the potential applications and considerations for utilising AI in chemical safety risk assessment, highlighting the importance of data quality, interpretability, regulatory compliance, uncertainty quantification, generalisation, and ethical considerations in the development and implementation of AI-driven solutions.
Overall, the workshop participants highlighted the potential of AI to revolutionise chemical risk assessment by addressing existing challenges, enhancing decision-making processes, and fostering collaboration across disciplines and sectors. However, the other publications related to AI and ML regulatory acceptance and readiness identify certain challenges that need to be addressed for the use of ML AI in chemical safety risk assessment based on similar parameters:
-
Data Availability and Quality: ML and AI models rely heavily on large datasets for training. However, obtaining high-quality chemical safety data, especially for rare or novel compounds, can be challenging. Additionally, data may be fragmented, inconsistent, or biased, which can affect the accuracy and reliability of ML and AI predictions
-
Interpretability and Explainability: ML and AI models often operate as “black boxes,” making it difficult to understand how they arrive at their predictions or recommendations. Lack of interpretability and explainability is a significant barrier to regulatory acceptance, as it hinders the ability to assess the reliability and validity of model outputs
-
Regulatory Compliance: ML and AI models used for chemical safety risk assessment must adhere to regulatory guidelines and standards. Ensuring compliance with regulations such as REACH in Europe or TSCA in the United States requires rigorous validation and verification of model performance and reliability
-
Uncertainty and Confidence Estimation: ML and AI models often struggle to quantify uncertainty and provide confidence estimates for their predictions. Lack of uncertainty quantification can undermine confidence in model outputs, particularly in scenarios where decisions have significant consequences for human health and the environment
-
Generalisation and Transferability: ML and AI models trained on specific datasets may struggle to generalise to unseen data or transfer to different chemical classes or contexts. Ensuring the generalisation and transferability of models across diverse chemical compounds and scenarios is essential for their practical utility in chemical safety risk assessment
-
Ethical and Societal Implications: ML and AI technologies raise ethical and societal concerns related to data privacy, algorithmic bias, and transparency. Addressing these concerns is crucial for building trust and acceptance of ML and AI-based approaches in chemical safety risk assessment among regulators, stakeholders, and the public
Overcoming these challenges requires collaborative efforts among researchers, regulators, industry stakeholders, and policymakers to develop robust methodologies, standards, and guidelines for the use of ML and AI in chemical safety risk assessment. Transparency, accountability, and continuous validation and improvement are essential principles to ensure the responsible and effective use of ML and AI technologies in protecting human health and the environment.
In conclusion, emerging NAMs like Artificial Intelligence, Machine Learning, and Organ-on-a-Chip demonstrate a moderate level of regulatory readiness. The publications identified (see Table 5) suggest that significant developments are underway. However, more work is needed for their full acceptance and standardisation in regulatory processes. The innovative nature of these NAMs holds great promise for transforming safety and efficacy assessments. Nevertheless, clear regulatory guidelines, validation frameworks, and consensus on interpretation standards are essential for their broader adoption.
6. The use of NAMs in regulation
To this point the report has presented the expert perspectives and opinions on the way that NAMs can be integrated into regulation, including the criteria that will need to be met by individual types and study and approach to be considered for use by regulators. In order for the goal of integration to be actuated, there needs to be some practical implementation of NAMs to prove that NAMs can achieve the regulatory requirements and to show where further improvements are needed. This chapter gives a brief overview of important case studies published in the literature that have used NAMs in a regulatory context. A more comprehensive list of case studies is given in Annex 12 of this report. This chapter also includes a brief examination of the progress of NAM integration into the regulators decision making process under EU REACH by examining decisions made since 2014 in two aspects of this regulation: Substance Evaluation and Annex XV dossiers identifying substances of very high concern (SVHC).
6.1. Published case studies
A theoretical discussion on the applicability of NAMs in regulation needs to be backed up with viable case studies to improve support for their use in the regulatory community. This section presents some selected case studies that have endeavoured to demonstrate how individuals and groups of NAMs can be used to reach regulatory decisions. In the workshop “Towards an Animal-Free Regulatory System for Industrial Chemicals”, ECHA has reinforced the value and importance of meticulously planned case studies to showcase the practical application of NAMs in chemical safety assessment without animal testing, thereby bolstering confidence in their efficacy (ECHA, 2023). Annex 12 provides a summary of a selection of case studies identified in the literature search.
Apart from in vitro genetic tests, only a few NAMs have been effectively integrated into regulatory decision-making processes, primarily within the cosmetics industry. The cosmetics sector is an example of how far collaborative efforts addressing the prohibition of animal testing in cosmetics and innovation can take us when it is based on established biological knowledge (e.g., key events that indicate the likelihood of skin sensitisation being provoked by a given substance) and advancements in technology (e.g., biologically inert materials for cell cultures) (Grimm et al., 2023).
Guideline No 497 on Defined Approaches (DAs) for skin sensitisation represents a first-of-its-kind product for the OECD (OECD, 2023a). The successful validation and regulatory acceptance of these in vitro assays for predicting skin sensitisation represent a milestone in the application of NAMs for chemical safety assessment, paving the way for reduced reliance on animal testing in this domain (Casati et al., 2022).
However, for data from NAMs to effectively inform regulatory decisions regarding chemical safety, it is crucial to understand levels of human exposure and the role of exposure information in different regulatory contexts (Westmoreland et al., 2022). NGRA, defined as an exposure-led, hypothesis driven risk assessment approach that integrates in silico, in chemico and in vitro approaches, provides a way to integrate new types of NAMs data from various sources into the decision-making process (Dent et al., 2018). Bury et al. (2021) apply the TTC principle for the safety evaluation of cosmetics ingredients when human exposures are sufficiently low and toxicity data are scarce. In their paper, Bury et al. illustrate the use of TTC as a pragmatic tool for safety assessment, focusing on case studies of cosmetic ingredients (Perilla alcohol, Basic Blue 124, Trifolium pratense flowers) with low consumer exposure. They demonstrate how the TTC approach can be effectively applied to ensure the safety of cosmetic ingredients for which toxicity data are scarce, especially when exposure levels are low.
A 10-step NGRA framework for applying RxA and NAMs in cosmetics safety assessment for use in cases where a TTC approach to cosmetics safety assessment is not possible, is described in Alexander-White et al. (2022). Several case studies were identified in our literature review that provide an application of the NAM/RxA framework, for the safety assessment of caffeine (Bury et al., 2021), propylparaben (OECD, 2020; Vandecasteele et al., 2021) and vanillin (Gautier et al., 2023). In the context of NGRA the utilisation of “omics” technologies has witnessed rapid expansion, particularly in deriving PoD values. A RxA approach applied to metabolomics data obtained from in vitro skin and liver models (RHE and HepaRG® cells) has been illustrated by Jacques et al. (2021). Focusing on the cosmetic ingredient DIV665, which has solely been investigated through in vitro assays, alongside a structurally analogous reference compound, PA102, previously assessed using traditional in vivo toxicity approaches, the study seeks to illustrate the potential for enhancing the safety assessment of cosmetics by integrating metabolomics data from in vitro models. A recent paper published by Silva et al. (2024) shows the use of transcriptomics data obtained from human liver spheroids exposed to Perfluorooctanoic acid (PFOA) combined with PBK modelling to derive a health-based guidance value for PFOA intake (ng/kg BW/day). The study demonstrates the combined utility of an “omics”-derived molecular point of departure and in silico quantitative in vitro to in vivo extrapolation (QIVIVE) workflow for setting health-based guidance values.
Furthermore, in the cosmetic sector, we have identified several case studies using NAMs to perform an ab initio NGRA (i.e. in vitro bioactivity assays to derive points of departure PoDs and physiologically based kinetic models combined with computer-based in silico predictions) to a hypothetical safety assessment under various exposure scenarios of cosmetic ingredients, i.e. 0.1% coumarin in face cream and body lotion (Baltazar et al., 2020), 0.1% coumarin in face cream and 1% in a non-spray deodorant (Reynolds et al., 2021); 1% Phenoxyethanol in body lotions (OECD, 2021). Middleton et al., 2022 describe a systemic safety toolbox and workflow based on the early-tier assays and models used in the above case studies, underpinned by the principles of benchmarking NAM data against traditional methods and assessing the utility of these tools within the context of risk assessment.
NAMs offer alternative approaches for extrapolating toxicological data from animal models to humans, addressing interspecies differences in sensitivity and response to chemicals. Human-relevant in vitro models, PBK modelling, and (Q)SAR modeling can provide more accurate predictions of human toxicity based on mechanistic insights and empirical data, reducing reliance on animal testing and improving the relevance of risk assessments.
A case study by Chen et al. (2023) presents a PBK modelling approach to predict acetylcholinesterase (AChE) inhibition, a common mechanism of acute neurotoxicity, in rats and humans after acute exposure to fenitrothion (FNT), an organophosphorus pesticide. By integrating in vitro data on fenitrothion metabolism with physiological parameters, the PBK model enables quantitative extrapolation from in vitro to in vivo conditions. The study demonstrates that the PBK modelling-facilitated extrapolation accurately predicts AChE inhibition in both rats and humans following fenitrothion exposure. The approach described in the paper claims a proof-of-principle for applying this approach in a 3R-based chemical risk assessment paradigm.
Algharably et al. (2022) published an example to evaluate to which extent drug-induced side effects or chemical-induced adverse effects could be quantitatively predicted starting with in vitro data, using three case studies (ibuprofen, amiodarone, and chlorpyrifos). The work highlighted important points to consider when comparing in vitro conditions to in vivo situations and showed the applicability of QIVIVE and that it can provide reliable results when compared against in vivo data.
NAMs are useful for prioritisation and screening, particularly in the context of chemical safety assessment. In a case study involving the food mycotoxin zearalenone (ZEN) and its metabolites Ehrlich et al. (2015) employed a combination of in vitro and in silico methods to assess the estrogenic potencies of ZEN and its reduced metabolites. The study showed the suitability of the model in evaluating the estrogenic potency of this group of compounds. However, the authors caution that while the present work suggests a relatively low concern for estrogenicity of ZEN oxidised metabolites, this finding alone is insufficient to definitively determine safety due to the potential involvement of other relevant and independent toxic mechanisms and MIEs. The approach outlined in this study holds particular relevance when rapid data acquisition is necessary for compounds that are not readily available commercially, unstable, or challenging to purify or synthesise.
A limitation of NAMs that might bring lack of consistency and uncertainty is illustrated in a case study by Sprenger et al. (2022) in which a battery of in vitro tests and omics techniques has been used to predict genotoxicity of pesticidal active substances (imazalil, thiacloprid, and clothianidin). Transcriptome analysis from animals treated with the three pesticidal compounds indicated genotoxicity in rat liver for clothianidin (CTD). In contrast, CTD was positive in two out of three in vitro clastogenicity assays and negative in a number of follow up in vivo tests. Computational approaches supported the assumption of a clastogenic potential for CTD. Whilst transcript signature of CTD in human HepaRG liver cells and Comet assay did not show a clear-cut classification of CTD as genotoxic or non-genotoxic. The authors used a WoE decision, considering in addition to the regulatory studies especially the findings in human cells and the extraordinarily high doses of CTD administered in the animal study used for the transcriptomic approach, they conclude that CTD does most likely not pose a genotoxic risk to consumers exposed to residues of CTD via the diet.
These case studies serve as examples of the real-world application of NAMs, showcasing both their potential and their constraints.
6.2. Assessment of the use of NAMs in regulatory activities (REACH) in the EU
True evidence of the integration of NAMs into regulatory activities can only be demonstrated by their use in actual decisions made in a regulatory context. To examine this, two regulatory activities within the EU REACH regulations were examined by comparing the number of NAM studies used versus the number of traditional in vivo studies.
6.2.1. Assessment of SVHC justification report (Annex XV reports)
Methodology: All substances examined as being a possible SVHC because of being a suspected endocrine disruptor or having “Equivalent level of concern having probable serious effects to human health (Article 57 f)”, meaning skin sensitisers were also included since 2014 was identified from the ECHA website (ECHA, n.d.) and the report justifying the decision was examined. Where a substance had an equivalent level of concern for both human health and the environment, only studies related to the human health aspect were examined. Each source of information stated in the report was assigned as either an in vivo study or a non-animal study. This concern was chosen as both endpoints covered by this description are expected to include both in vivo and in vitro studies
Discussion: The assessment identified 22 substances as meeting the criteria for inclusion in this assessment (see Annex 13 for full details). The breakdown of the studies used in the assessment across all substances were as follow:
-
108 epidemiological studies were used, exclusively for skin and respiratory sensitisers
-
362 in vivo studies
-
316 non-animal studies of which 6 were in silico studies using various databases
The endpoints chosen for assessment were because they are the endpoints that either have a defined approach to use NAMs to address the endpoint or have a component of their definition that would require a NAMs to assess. This snapshot shows that even for endpoints where NAMs should be expected to be used extensively, the use of animal study results is still very important. Much of this will be from historic data. The use of epidemiological data is very important for endpoints closely related to occupational health. The evaluators are using in silico data to support the assessment but the choice of the data source is not consistent and the reason for choosing one source over another was not clear. It was not apparent that multiple in silico sources were used which goes against the recommendations from the literature. Finally it was not immediately apparent whether one type of data was given greater weighting in a final conclusion than another. An interesting extension of this work would be to identify whether there is a movement towards more in vitro studies being used over time as NAMs approaches gain greater regulatory readiness.
6.2.2. Substance evaluations since 2014
Methodology: The ECHA website page on historic and ongoing substance evaluations was used as the data source. All evaluations that were started from 2014 onwards were examined. Only substances with a decision requiring further information were assessed in this survey. All decisions (including where two or more rounds of decisions were reported) were assessed and each individual study assessed as to whether they were an in vivo or NAM (in vitro, in chemico) study. The NAMs were further categorised because a significant number of studies were on fish, amphibians, molluscs or other invertebrates. Although these are not “animals” from a UK law perspective they would be regarded as animals in other jurisdictions so whether they are non-animal methods is inconclusive. Where options of multiple studies were given to the registrant, each study was included in the assessment.
The studies were also categorised by the endpoint for which each study was intended to address. Several substances had different studies required to address different concerns within a single decision, so certain substances will be double counted in the “No. of substances” column in the results. It was also noted that the endpoint examined was not always the endpoint for which there was an initial concern that prompted the evaluation (for full details see Annex 14).
Discussion: A summary of the finding of this assessment is shown in table 6.
It is important to note that a PBT assessment includes consideration of persistence, which is generally assessed by use of biodegradability studies which are always in chemico studies. This means that the number of studies that do not require animal studies for this assessment is inflated compared to the other endpoints. As this is an environmental endpoint, bioaccumulation and toxicity studies are performed on fish, molluscs or other sediment dwelling organisms depending on the environmental compartment that the substance is expected to be found in the highest concentration. Perhaps unsurprisingly the endpoints that require NAMs are those either with a defined approach (skin sensitisation) or whose definition require knowledge of the biological mechanisms involved (endocrine disruptor). It should be noted that where animal studies or potential animal studies were required, either fish or amphibians were often the target organism. The reason for the choice of target organism was not always obvious from the decisions. The decisions requiring in vivo studies for skin sensitisation were made before the defined approach for this endpoint was published, so it can be expected that such studies will not be requested in the future. Perhaps the most striking result to note was that in vivo studies are almost always requested when the endpoint of concern is carcinogenicity, mutagenicity or toxicity to reproduction. The in vitro studies required were the fully validated mammalian cells that have been a requirement of REACH from its implementation and would generally not be regarded as New Approach Methodologies. It may be surprising that these studies were not already in the registration dossier of the substance and the requirement made part of a dossier evaluation instead. It may be that the substance had not been registered on sufficient tonnage to trigger this obligation. Once this is taken into account, it becomes apparent that NAMs for these endpoints are not regarded as giving the same level of confidence as in vivo studies. The evaluators do not appear to have considered the use of a RxA strategy to address these endpoints in most substance evaluations, with most consideration applied when a RxA position was included in a testing proposal by the registrant (see report below).
ECHA published the report titled “The use of alternatives to testing on animals for the REACH Regulation” in 2023 under their obligation from Article 117(3) of REACH that presented how NAMs have been used in REACH registrations. It reported that across 6,290 substances assessed:
-
89 % contain at least one endpoint in the dossiers where an adaptation or other argument was provided instead of a study result
-
63 % contain at least one RxA adaptation
-
43 % contain at least one WoE argument
-
34 % contain at least one (Q)SAR prediction
More detailed analysis showed that RxA was largely used to satisfy the higher tier mammalian toxicity endpoints such as repeated dose toxicity, whereas (Q)SAR was largely used to predict ecotoxicity endpoints. When discussing substance evaluation, the report emphasised the use of RxA to justify the use of grouping to reduce the number of new animal studies that needed to be commissioned. They highlighted that the recent adaptation to use RxA had been accepted for acetalisation products between glucose and long chain alcohols, and some organic carboxylic acids in the form of their sodium salts. As with the assessment of Annex XV dossiers, an interesting project for the future would be to identify whether there is progress to an increased requirement for new NAMs methods compared to new in vivo studies over time.
7. Perspectives on the use of NAMs in risk assessments related to food
NAMs are progressively supplanting traditional animal models in toxicity prediction, emphasising mechanistic data over conventional endpoint measurements derived from animal studies. However, our literature findings have shown that the integration of NAMs into regulatory frameworks remains limited, primarily observed within the cosmetics and chemicals sectors. This chapter gives a summary on the use of NAMs for food safety assessment reported in the literature
In food safety evaluations, global regulatory bodies such as EFSA (EFSA, n.d.), the U.S. FDA (Predictive Toxicology Roadmap), and the UK FSA (UK FSA, 2023) are acknowledging a paradigm shift and embracing newer tools for risk assessment, but at a slower pace than other sectors. A review by Reddy et al. (2023a) states that regulatory guidance on food safety testing in many jurisdictions still relies on data obtained from animal studies, mainly due to the absence of rigorously validated models capable of accurately predicting systemic toxicity, which is a prerequisite for establishing safety reference values for food and food ingredients. The authors reviewed the current regulatory landscape surrounding the use of alternatives to animal testing in general, discussing several factors and barriers influencing the regulatory decision on alternative models, such as the complexity vs. relevance debate (while in vivo models are often perceived as more complex and superior, some researchers emphasise the relevance of test models over their complexity in predicting substance safety) and the need for developing performance-based standards, particularly in areas like repeated dose toxicity, carcinogenicity, and reproductive toxicity. In a subsequent review, Reddy et al. (2023b) provide an overview of the most recent alternative methods available for toxicity testing. These encompass in vitro approaches (such as Caco2 cell lines, primary hepatocytes, organ-on-chips, Bhas 42 cell lines ) computational (including (Q)SAR, read-across, PBK, TTC) and non-mammalian models (e.g Zebrafish and C. elegans), alongside AOP, omics and IATA, detailing their application in evaluating food safety. The authors note that while some of the in vitro and in silico tools contribute to WoE assessments, TCC is used by EFSA and JECFA for evaluating flavouring substances and by the FDA for food contact materials, non-mammalians models, omics and IATA are presently not integrated into regulatory food safety evaluations. EFSA has developed, calibrated, and validated generic PBK models using case studies for chemicals pertinent to food safety. Moreover, EFSA introduced “TKPlate” in 2023, an online platform facilitating scientists and regulators to model and predict toxicokinetic (TK) and toxicodynamic (TD) properties (Dorne et al., 2023).
Punt et al. (2018) conducted a survey among toxicologists from academia, industry, and regulatory authorities to gather expert opinions on the adoption of alternative methods in food safety evaluations, with a particular focus on formulating suggestions to promote the acceptance of non-animal methods, especially concerning kinetics. The study identified three primary barriers: the uncertain predictability of 3R methods and the lack of validation, insufficient guidance from regulators and industry, and the absence of harmonisation in legislation. The recommendations that emerged from this study included diverging from rigid regulatory data requirements, establishing funding opportunities for the validation of non-animal methods for kinetics and developing guidance documents.
Blaauboer et al. (2016) provide an overview of the components considered in current safety assessments for food and food ingredients. The authors emphasise that while safety evaluations for food additives typically adhere to established protocols, assessments for complex foods and ingredients are often more tailored to specific cases. They discuss the incorporation of new methodologies into safety assessment practices, particularly focusing on mechanisms of toxicity, understanding internal exposure kinetics, and employing modelling techniques. The authors also outline a step-by-step approach for conducting a comprehensive assessment, which includes exploring potential exposure scenarios, examining internal exposure kinetics, evaluating target-specific toxicities, investigating toxicity mechanisms, conducting in vitro and in vivo evaluations, and ultimately balancing the benefits against the risks of adverse effects.
Omics have been suggested for the use in food safety assessments by a number of authors. Levorato and Nathanail (2021) discuss the application of toxicogenomics to perform high throughput screening to assist safety assessment of chemicals in food. Su et al. and Zhou et al. discuss how different omics can be used to support food safety assessment but address related areas also: food authenticity and optimisation of natural colourings respectively.
Other regulatory jurisdictions, such as Japan and China, are also increasingly acknowledging the significance of non-animal approaches and actively exploring alternatives within the food sector, as presented in the 2021 International Workshop for Non-Animal Approaches in the Food Sector held in Japan (Ohta et al., 2023). For instance, the International Life Sciences Institute (ILSI) Japan is conducting the Alternative Animal Testing (AAT) Promotion Project in Food Sector, comprising four working groups engaged in activities such as predicting internal exposure, using existing data and information effectively, generating and disseminating case reports, and organising international workshops and symposiums. Similarly, the China National Center for Food Safety Risk Assessment (CFSA) is at the forefront of developing Non-Animal Methods for NGRA in China’s regulatory food safety assessment. The plan is to accelerate research and development in hazard identification technologies, focusing on human-derived cells and zebrafish in vitro models. It also involves making full use of modern toxicological technologies like genomics, transcriptomics, proteomics, and metabolomics, coupled with comprehensive data integration and analysis, to uncover toxic pathways and elucidate toxic mechanisms. Furthermore, efforts include enhancing the hazard assessment system through methods such as structure-activity analysis, PBTK-QIVIVE, and promoting the construction of efficient platforms like laboratory intelligence and digitisation to effectively support national food safety implementation strategies (D. Yang et al., 2023).
8. Conclusion and next steps
New Approach Methodologies must play a key role in future regulations if the 3R’s goals expressed by governments and organisations across the globe are to be achieved. This role is likely to transform from a supplementary role where they can achieve two of the 3Rs, reduction and refinement, of animal studies in the near future to the other one of the 3Rs, replacement, in the medium to long term. It is very unlikely that this evolution will occur at a uniform rate across all toxicological endpoints. NAMs have already replaced animal studies where endpoints examine effects on a single organ and biological mechanisms are well understood (skin irritation, skin corrosion, eye irritation, skin sensitisation). However, as the endpoint becomes more complex, addressing multiple organs over extended periods of time, the rate that NAMs will replace animal studies will reduce. This is especially the case where animal studies are regarded as being well established and risk assessors are comfortable in making decisions based on them. The other biological effects where NAMs will play more than a supporting role are for emerging adverse effects where there is not an established animal study to replace, such as developmental neurotoxicity or immunotoxicity. In the absence of established benchmarks from animal studies, it may be that research into these endpoints will have animal studies and NAMs running in parallel. It is not clear how and when regulations would address these emerging risks, but it is clear that different regulators are actively identifying and monitoring this issue.
Even the acronym NAM triggers different opinions as to the best term for which it should be used for, with each alternative resulting in potential conflict between different regulations in different regions. It may be that trying to introduce the term into regulations as a legally binding expression could be prohibitively problematic and it should remain as a useful descriptive term to communicate with non-experts in the field, however movement towards a common understanding of its meaning is necessary to avoid misunderstandings across the toxicology community. One description that has universal acceptance is that NAMs are technologies that will achieve the 3Rs goals.
Recommendation: Despite its short-coming, New Approach Methodology is becoming the established term used to describe methods that will achieve the 3R goals. It is an important totem around which a wide range of research can be described and integrated. Care should be taken around inclusion of the term into legislation as agreeing a definition could be a lengthy process.
Investment in research into NAM development is thought to be growing, with the EU and US at the forefront. Although there are some high profile publicly funded projects such as PARC in the EU, much funding can come from sources that would not be immediately associated with NAM research, for example from regional partnership programmes or from research into new materials which incorporates an element of toxicology research. It is felt that maintaining the approach to use both large scale broad projects alongside smaller focused projects is the best approach. For the projects to have the most influence it is important for the project management to ensure the whole project maintains a focus on the goals of the project and to work within a reporting framework. This would ensure that the applicability of any deliverables can be understood by regulators and that any data provided is Findable, Accessible, Interoperable, and Reusable (FAIR). Whilst many EU funded projects include a commitment to FAIR data, it could be useful for researchers in these publicly funded projects to include regulators in their stakeholders or advisory board and to include an assessment of how the results of the project have advanced the potential for use of NAMs in the regulatory arena, including the following factors
-
A clear description of the biological and regulatory endpoints for which the research could be used
-
An assessment of the regulatory readiness of the NAMs included in the project. This should be made for each of the different regulatory processes, e.g. screening, hazard and risk assessment
-
Commentary on where the NAM would fit within a battery of NAMs, for example does it fill a gap in an existing AOP or is further research into other KEs in an AOP needed before the NAM can realise its potential
-
Full transparency regarding uncertainties in the NAM (or battery of NAMs), following a standardised template if one is available
This would help achieve the goal of training both regulators in new methods and researchers in the criteria that their new technologies must meet to reach regulatory acceptance. Whilst other global regions are taking an active interest and have very active research groups, from a regulatory perspective they are generally waiting to see the direction that the US and EU will take before taking a firm position themselves. Currently NAMs can be used in regulatory submissions but uncertainty in the quality and relevance of the conclusion from these studies means that there is a common consensus that regulators are reluctant to reach decisions without any in vivo data available. Examination of recent regulatory decisions in EU REACH appears to support this belief. The exceptions to this position are where the generation of new in vivo data is prohibited, for example for cosmetics in the EU.
Recommendation: Where the UK FSA is involved in funding or monitoring research, a requirement should be placed on researchers to address the criteria needed to make the transition from research to a regulatory tool.
So far we have discussed the approaches that researchers should take to improve the regulatory confidence in NAMs. However, there needs to be changes from both sides to facilitate the greater integration of NAMs into regulatory decisions but this may depend on the regulation and region where the regulator is based. In the US, regulators such as the EPA and FDA both fund and perform a significant proportion of the research into NAMs meaning that they can be expected to have a deep understanding of the potential and shortcomings in the research. As the US regulators have the primary responsibility to undertake risk assessments under many of their regulations, it is logical that they would be proactive in the development and assessment of NAMs. Regulations in other regions, such as REACH in the EU, place the initial responsibility of risk assessment on industry, with the regulators ensuring that the registration is done in accordance with the wording of the regulation and that the conclusions are scientifically justified. Under this style of regulatory framework, neither industry nor the regulators will have as much incentive to move away from an established paradigm that has legal precedent, especially where they have not been involved in the development of the alternative approaches. Some regulators actively engage in research, while others take a more passive approach. This discrepancy may stem from the structure of different regulations. Regulators with primary responsibility for risk assessment have greater flexibility to use NAMs to support or replace animal studies. In contrast, risk assessors for regulations where the industry is responsible for the initial risk assessment seem more hesitant to accept NAMs data, even when the regulations permit it.Regulations that are designed to understand and classify the hazard of a substance will often need to use animal study results because the globally accepted approach to classification is based on the results of animal studies. There would need to be a paradigm shift in the approach to this regulation. Although some stakeholders are advocating this move to a NGRA, it is unlikely that this will be seen in the short to medium term. The EU-funded PARC project is currently examining the possibilities offered by a new approach to risk assessment but it will not conclude until 2029. In the short-term the most effective way to reduce new animal studies is to make use of existing data, for example by the wider use of RxA. This approach is well established in regulations already but indications from REACH suggest that the quality of RxA in REACH registration dossiers is inconsistent. This may be in part due to insufficient guidance, particularly an absence of case studies which demonstrate both good and poor approaches. There is a wide range of academic literature that suggests ways to justify RxA but it is unclear whether this is being transferred into guidance documents that industrial registrants can easily access and understand.
Some commentators expressed an opinion that a barrier to NAM adoption is that risk assessors are more comfortable with making decisions based on animal studies both due to greater familiarity with them and confidence in legal precedence established in their use. A change in this mind-set might be achieved by a small change in the approach to assessing testing proposals both from registrants and from risk assessors during an evaluation process. Within the REACH authorisation process under REACH, an applicant needs to fully assess any alternatives to the use of an Annex XIV substance and in some cases explain why they do not meet the technical requirements that the Annex XIV substance does. If this approach were to be taken for new animal studies, whereby alternative approaches using NAMs were demonstrably considered in decisions, even should the NAM (or battery of NAMs) not provide the quality of data that an animal study would, the deficiencies would be transparent and future research could be directed towards overcoming the issues. It would also require the risk assessors to become more familiar with alternatives and for them to better understand the opportunities and shortcomings associated with them.
Recommendation: The use of NAMs to justify read-across or weight-of evidence conclusions may be the most effective approach to reduce and refine animal testing in the short to medium term. The UK FSA should consider the requirements of the legislation for which they cover and make consideration of how to use existing data a defined step in their decision-making process. Where a decision to commission new animal studies is made it should include a transparent assessment of why NAMs do not provide the necessary information alongside the justification for why the selected animal study is appropriate (NB. this might be more relevant to other regulatory authorities other than the UK FSA).
Another barrier to widespread NAM adoption is thought to be that using a battery of NAMs in place of a single animal study would be considerably more expensive. In addition, it is not clear whether they would be as available from contract research organisations as animal studies. Although some large companies are very active in developing and using NAMs to guide their innovation decisions, partly because they are able to perform much of the work in-house, small and medium-sized enterprises (SME) would probably need to use external laboratories and experts. If there was limited availability and higher costs, it could make innovation from these companies very difficult to achieve. Should the EU-based drive for companies to incorporate the safe and sustainable by design (SSbD) into their innovation practices, it can be expected that there will be an increase in NAM generated data. There needs to be clear guidance on how this data might also be used to support regulatory submissions.
Recommendation: An examination into the decision-making process used by CROs to decide which studies they offer commercially could clarify the steps that are needed to encourage a wider range of them to offer NAMs. Once there is competition in the market to offer NAMs, the price of a battery of tests should become more comparable to that of an animal study.
The advocates for SSbD should demonstrate how data generated during the innovation stage could be re-used for subsequent regulatory submissions. This could be supported by the large companies active in this area by continuing to present case studies.
For the specific area of food safety, NAMs are already being used in the regulatory decision-making process in different global regions. Most NAMs can be effectuated in a short period of time compared to an animal study, which makes them vital when immediate decisions need to be made, for example to assess the impact of food contamination. EFSA is very active in producing guidance documents to allow stakeholders to clearly understand where and how different types of NAMs can be used in the regulations that they enforce. In the US, the FDA are playing an active role in projects examining the link between PoD measured by animal studies and those from NAMs such as ToxCAST, and assessment of emerging issues such as alternative protein sources. Although smaller regions may not be able to commission wide ranging projects, they are seeking to collaborate with international research and do undertake tightly focused research (e.g. Health Canada examining grouping for the risk assessment of microbial consortia). One of the unique features of food safety that may lend itself to integration of NAMs is that many of the substances of concern are not intentionally present. Food can contain degradation products of pesticides and biocides used in both farming and processing, metabolites from bacteria, fungi and other organisms and unintentional contamination from food packaging materials. These substances may be complex in structure and composition, and be present in only low concentration making their identification and isolation difficult. They are likely to have very little existing toxicology data, both animal and non-animal studies. This means that animal studies and NAMs have the same “starting point”, so there is not such an established preference for the use of one approach ahead of the other as there might be with bulk chemicals. In these situations, NAMs can be developed in parallel with animal studies where the animal studies are designed to support the use of the NAM, rather than the other way around which is the short term goal expectation for most NAMs in other regulations. This means that food safety regulators could play a leading role, alongside those addressing cosmetic products, to collaborate with and increase the confidence of regulators and risk assessors in other sectors.
The UK Food Standards Agency published a roadmap towards the “Development, Endorsement and Regulatory Acceptance of New Approach Methodologies (NAMs) in Chemical Risk Assessment and Beyond” in 2023. In this roadmap, a commitment to the greater integration and use of NAM technology into risk assessment was made, with an emphasis on the use of digital technologies and data handling. The amount of data generated by some NAMs such as omics can be very large, possibly out of the scope of humans to properly synthesise the data into information, so the development of digital tools such as artificial intelligence and machine learning will be essential. The resources that are needed to develop such tools and databases can be great, so the adaptation and use of existing tools would be a sensible approach to achieve this goal. The roadmap emphasises the need to break down silos and for collaboration across sectors both nationally and internationally. As the toxicity of a chemical does not change geographically, data generated in any region would have relevance. Therefore, integration with existing databases and associated digital tools would be a cost effective way to achieve these goals. The specific requirements of food safety, for example the route of exposure, low exposure doses and data poor substances would mean that care should be taken to choose relevant tools, possibly through collaboration with other food safety regulators (e.g. EFSA, US FDA). The UK is at the forefront of research into NAMs both in academic research and industry, and its regulatory experts have a strong reputation internationally, so there should be ongoing active investment into the field to maintain this position both through UK funding and involvement in international projects.
The term NAM covers a broad spectrum of methods that are suited to different purposes in risk assessment, both in isolation and as part of a battery of tests. The roadmap emphasises the need for the UK FSA to clearly define the problem space that they intend to address. It emphasises a transition from the historic 3Rs approach (replacement, reduction, and refinement of animal experiments) to an expanded 6R principle that includes reproducibility, relevance, and regulatory acceptance using a workflow similar to the IATA approach being explored by the OECD. This will be fundamental to achieving accurate and efficient risk prediction by identifying existing NAMs that can be used, whether NAMs need to be used in conjunction with existing animal studies, or whether NAMs are needed to guide the design of any necessary animal studies as efficiently as possible. An example of this might be where a contamination issue arises that requires an immediate response regarding potential acute toxicity to consumers, screening using (Q)SAR could provide an immediate assessment of the potential severity. By clearly defining the goals of any assessment, the ability to define the most appropriate NAMs and how these should be integrated with each other, with existing data and with new in vivo data should be more easily facilitated.
To meet the goal of predicting risk accurately, regulators must define the required level of accuracy for assessments. A priority area should be addressing uncertainty, both for individual NAMs and batteries of NAMs, to ensure that the risk assessments are as precise and reliable as possible Although commentators have emphasised the importance of understanding the uncertainty around new methods, there is little guidance on how it should be measured, how uncertainty in different methods can be compared and contrasted, how uncertainty of batteries of tests should be examined and how much uncertainty is acceptable for different regulatory purposes. Progress towards a standardised approach to measuring uncertainty for NAMs and the ability to assess this against a benchmark of uncertainty of in vivo results will be essential for risk assessors to gain confidence.
Recommendation: The UK FSA may have the opportunity to integrate NAMs into their decision-making processes at a quicker speed than other UK regulators. The goal of cross departmental collaboration in the UK will allow the UK FSA to identify the opportunities to overcome barriers that regulators of other sectors believe restricts them using NAMs.
Regarding individual types of NAMs, there will be two hurdles that must be overcome to allow the use of NAMs for more than very specific endpoints.
-
Most commentators agree that in the short to medium term, NAMs will be supportive of animal studies for decision making. For this to move from explaining results in new animal studies to supporting the use of existing data from similar substances (e.g. grouping and RxA) there needs to be a clear link demonstrated between in vitro and in vivo dosimetry. This will be achieved by the development and validation of PBK models that will allow IVIVE to be made. A clear emphasis on the inclusion of PBK models or a full assessment of toxicokinetics in any AOP that addresses systemic toxicity in particular could help drive the development of these models
-
The evolution of linear AOPs to a network of AOPs will also be important to address complex systemic endpoints. The ongoing development of omics and organ-on-a-chip technologies, in association with artificial intelligence to process the quantity of data produced, will play a key role in this transition. One goal of these methods should be to demonstrate how different stages of an AOP interlink. For example, it is not immediately obvious to stakeholders other than experts in the area how the different omics technologies would be structured into a multiomics approach and whether all omics or only selected ones would be needed for a regulatory assessment
An aspect of risk assessment where omics and PBK modelling are expected to play a useful role is in the identification of low toxicity or “non-toxic” substances.
Recommendation: PBK and IVIVE will be intrinsic to any NAM-based risk assessment and NGRA. The UK FSA should continue their activities in this area and place a priority in identifying and monitoring the development of models that are directly relevant to food safety, such as gut models. As the trade in food is international, this might be best done through collaboration with similar organisations in other regions (e.g. OECD, EFSA, US FDA, Health Canada). Such collaborations should proactively engage with smaller countries who may not have the resources to be able to monitor developments by themselves. This should increase confidence in food imported into the UK.
The UK FSA should use their influence to promote the inclusion of PBK aspects into all AOPs and associated IATA that address systemic endpoints.
The UK FSA should continue its close collaboration with researchers in omics, encouraging them to consider how an individual method can be incorporated into a multiomics approach to risk assessment as part of their research.
Validation is regarded as an essential aspect to give regulators confidence in the results of NAMs. Unfortunately it is also regularly identified as a significant barrier to NAM integration. Validation demonstrates that the results of a NAM are reproducible, the biological relevance is proven and the uncertainties are clearly described. The current process of validation through the OECD, which is well established for new in vivo studies, has been described by stakeholders as long and open to political considerations. The funding for the interlab ring trials can be uncertain and sometimes relies on the goodwill of participants. This approach is currently applied to in vitro methods but there is a fear that the lengthy procedure and rapid developments in the area could mean that when a method is validated, it will have been superseded by other methods. Some stakeholders have expressed a view that both omics and organ-on-a-chip technologies could also be validated by the existing approach but others comment that the validation process for these methods would take even longer than for in vivo and in vitro studies. The US also uses qualification of methods which appears to be a less strict approach to validation than the OECD validation process, and is used for activities such as screening and prioritisation.
In silico methods do not go through the same formal validation process but regulatory guidance often requires that (Q)SAR methods should be “validated”. This generally means that the user needs to make the assessment on the validity of a model for the substance and endpoint under assessment. There is a lot of literature on the criteria that should be assessed to make this judgement but this relies on the relevant information being easily accessible for a non-expert user both when choosing a method and when assessing the results.
Recommendation: The UK FSA could develop a template for the assessment of in silico methods that is tailored to their requirements to ensure consistency in the acceptance or rejection of different tools across the organisation. This could be designed to account for the type of assessment (e.g. screening for rapid decision making could have a lower bar for validation). They could also use their influence with international bodies and researchers to ensure the criteria for validation are clearly stated for each model, especially in toolboxes and databases where multiple tools can be found.
Although validation is the accepted benchmark for validation currently, the FSA could examine the concepts of “endorsement” and “qualification” that are starting to be used in other regions for some NAMs.
As previously stated, it is expected that multiple NAMs will be used to achieve the risk assessment for an adverse event where a single animal study is currently used, although perhaps not by generating exactly the same data. When designing this battery of tests, it is expected that an AOP will be used to generate a hypothesis for which an IATA will be used to identify the tests needed. As AOPs are substance-agnostic and the route through an IATA could vary from substance-to-substance, performing a validation in the way that an individual in vivo or in vitro study is validated (e.g. ring trials etc) may not be the most realistic approach. However, it is essential that confidence in an approach can be quantified in some manner to ensure acceptance across different regulatory jurisdictions. The ongoing work in the OECD around AOPs and IATA will be key for this. Some stakeholders expressed the opinion that whilst IATAs give the flexibility in research to address a wide range of substances and endpoints, some will eventually evolve into defined approaches that are more suitable for some regulatory requirements (e.g. hazard assessment and classification) as seen for the skin sensitisation endpoint. A formal framework for this evolution could allow researchers to structure their IATAs to facilitate this transition more efficiently.
Recommendation: The UK FSA should identify which of their tasks could be satisfied using an IATA approach and which would require a defined approach by defining their needs and problem space (as stated in their roadmap).