
Executive Summary
Whole-genome sequencing (WGS) for food-borne disease (FBD) surveillance provides many benefits, including new insights in disease transmission, virulence and antimicrobial resistance (AMR), fast and precise outbreak tracing and source attribution, as well as streamlined and reproducible analysis through digital data that, from a technical point of view, can be easily shared. The National foodborne disease genomic data platform (the PATH-SAFE platform) will offer a trusted environment for WGS data sharing and analysis for UK agencies involved in FBD surveillance.
Following the successful implementation of the platform for Salmonella, in the second phase the platform will be expanded to Escherichia coli and Listeria monocytogenes. Where possible, the platform will draw on existing and validated solutions. For de novo genome assembly, EToKI and vanilla SPAdes provide the best results for E. coli, and Pathogenwatch provides the best results for L. monocytogenes. Analysis of genomic data is greatly enhanced by assigning genomes into well-defined cluster groups, which should be available on the PATH-SAFE platform. Specifically, tools for MLST and cgMLST should be available on the PATH-SAFE platform for both E. coli and L. monocytogenes. In addition, HierCC and ClermonTyping tools should be available for E. coli. The platform should implement tools for clustering E. coli and L. monocytogenes based on MLST/cgMLST profiles. Clusters should be named according to their HierCC codes. The PATH-SAFE platform should implement a tool for predicting E. coli serotypes from sequence data. ECTyper has been selected as the only up-to-date tool for this purpose. Although serotype determination of E. coli isolates is useful for historical reasons, the platform should be designed in a way that makes it easy to switch to hierarchical clustering of isolates.
The identification of genetic virulence determinants is essential in the analysis of E. coli and L. monocytogenes. VirulenceFinder and AdhesiomeR should be implemented in the PATH-SAFE platform for virulence determinant identification in E. coli. The Pasteur L. monocytogenes Scheme has been selected for virulence determinant identification in L. monocytogenes, although none of the available databases seems to include all known genes determining virulence in L. monocytogenes. As the PATH-SAFE platform is expanded to new food-borne pathogens, integrating a tool to differentiate species will be useful. Speciator has been validated and shown to be 99.9% in agreement with Kraken in correctly assigning E. coli genomes.
The number of metadata fields should be small initially to facilitate upload of data and use of the PATH-SAFE platform and to be consistent with UK GDPR obligations. Minimum metadata requirements of the platform should be compatible with the metadata collected by each agency. They should also be compatible with concerns around data sharing and legal obligations. All metadata must be processed in line with UK GDPR guidelines and align with organisational policies and relevant legislation. The PATH-SAFE platform should implement a gated access model that will allow participating agencies to share additional metadata with trusted partners and at the same time minimise the risk of leaking sensitive information.
The PATH-SAFE platform should have an automated QC mechanism for validating uploaded metadata. Options for both bulk upload of metadata and for upload of individual metadata fields should be offered by the platform. In addition, functionality for regular automated uploads could be provided.
Experiences with implementing WGS for FBD surveillance in the UK, Switzerland and Canada show that collaboration of reference laboratories carrying out sequencing analyses and epidemiological and One Health units providing metadata is critical for prioritising isolates for outbreak investigations.
Contributors
Abbreviations
1. Background & objectives
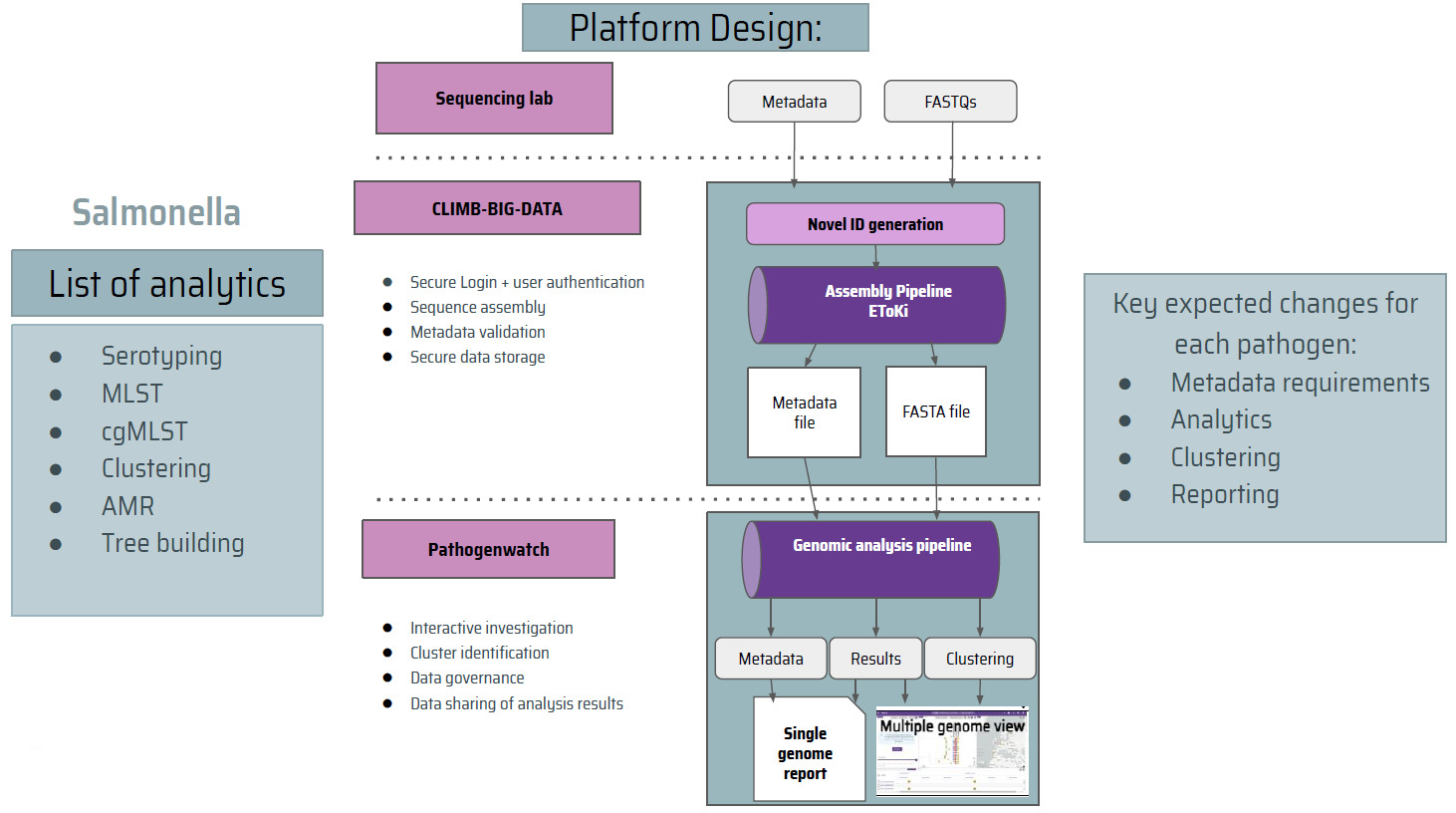
The PATH-SAFE consortium is developing a platform for UK agencies involved in foodborne pathogen surveillance to share pathogen sequence data. The platform operates by processing decentralized sequencing data by participating agencies. The data includes genomic sequences and contextual metadata and these are analysed and converted into visual reports for end-users. Reports can either cover a single genome or provide a multi-genome view, helping end-users identify clusters of isolates for further investigation. The PATH-SAFE platform’s flexible design allows for easy adaptation to new pathogens, though each pathogen may require adjustments to its metadata fields and reporting structures.
The platform will have a gated access model, meaning that only designated personnel from participating UK agencies involved in foodborne disease surveillance will have access. The platform is not a research platform but a public health/surveillance platform. This access model will minimise the risk of leaking sensitive information and allow agencies to share more data than on a publicly accessible platform.
After building the platform for Salmonella enterica as an exemplar pathogen, this phase of the PATH-SAFE project will deliver platform functionality for Escherichia coli and Listeria monocytogenes.
Differentiation of Shigella and E. coli is out of scope of this phase of the project.
2. Technical aspects of genomic surveillance of E. coli and L. monocytogenes
This section covers evidence-based recommendations on the most suitable pipelines and analysis frameworks for E. coli and L. monocytogenes. The same set of evaluations were carried out previously for Salmonella (PATH-SAFE, 2024b). The recommendations are not necessarily about which tool is best, but rather on how tools support the interpretation of genomic surveillance data and how they can help analysts meet technical and legislative requirements. The recommendations are deliberately not prescriptive but allow for the integration of multiple validated, curated and comparable solutions alongside each other. The recommendations will ensure flexibility of the platform with a view to future scale-up and technological and research advances, for example, increased use of long-read and hybrid sequencing platforms.
Validation reports for functional tools that are part of the PATH-SAFE platform are included in Appendix 1.
2.1. Assembly pipeline and quality control metrics
As part of the PATH-SAFE consortium, FSA has commissioned the University of Warwick Bioinformatics Research Technology Platform to evaluate and compare different assembly pipelines for Illumina short-read sequence data of E. coli and L. monocytogenes (PATH-SAFE, 2024a).The same set of assembly pipelines was tested as previously for Salmonella: SPAdes and the SPAdes-based assembly pipelines (EToKi, Pathogenwatch, and Unicycler), as well as, IDBA, MaSuRCA, MEGAHIT, SKESA, and Velvet. An assembly pipeline here refers to an assembler with different parameters for upstream and downstream processing steps.
A three-pronged approach was used to evaluate different assembly pipelines. First, representative read sets from the National Institutes of Health (NIH) sequence read archive (SRA) were assembled using all assembly tools in the test set, and the quality of results was compared using assembly metrics generated by Quast (Gurevich et al., 2013). Important quality metrics capture the number and length of assembled contigs, the number of called bases, genome completeness, the proportion of failed assemblies and the variance of these metrics when repeatedly assembling the same test data.
In a second approach, assemblies were further assessed for completeness of core genes and alleles using PubMLST. Completeness of L. monocytogenes core genes was evaluated using a new cgMLST scheme shared by Institut Pasteur (Moura et al., 2016). The third approach compared assemblies generated by approach 1 to reference strains E. coli K12 MG1655 and Listeria EGD-e.
All three approaches show that EToKI and vanilla SPAdes provide the best results for E. coli, and Pathogenwatch provides the best results for L. monocytogenes (PATH-SAFE, 2024a). In general, SPAdes-based pipelines consistently perform best followed by MEGAHIT (PATH-SAFE, 2024a). Older assemblers, for example, Velvet and SKESA, are mainly of interest as reference points for backward comparisons, but are unlikely to be used in modern assembly pipelines.
All comparisons were conducted using short-read sequence data. Long-read sequence data are expected to quickly become more important as prices will decrease. Evaluation of assembly tools may need to be repeated with long-read data. This evaluation is not on the deployment schedule for the current release of the PATH-SAFE platform but will be considered in the next release. In this context, we note that Pathogenwatch does not treat long-read sequence data differently from short-read sequence data. Pathogenwatch runs all analyses on genome assemblies and is agnostic to the method by which they were produced.
Recommendations:
-
Genomes assembled by the SPAdes assembler, as implemented in pipelines that incorporate suitable pre- and post-processing steps, including EToKi, Unicycler or Pathogenwatch, could all be accepted within the PATH-SAFE platform.
-
EToKi and vanilla SPAdes performed best when assessed for genome assembly quality and core gene assembly of E. coli.
-
Pathogenwatch was found to generate the best quality assemblies for L. monocytogenes.
-
Older assemblers, such as Velvet and SKESA, could be integrated in the PATH-SAFE platform as reference points for backward comparisons.
-
The PATH-SAFE platform will be built in a way that will facilitate the analysis of long-read sequence data in future developments.
2.2. Analysis framework
In a survey among end users of the PATH-SAFE platform, the following were all identified as essential analyses to support E. coli surveillance; lineage identification, clustering, serotyping, AMR genotyping, identification of virulence determinants, and identification of Shiga toxin producing genes. Similarly, lineage identification, clustering, AMR genotyping, and identification of virulence determinants were named as essential analysis for L. monocytogenes surveillance. Table 1 gives an overview of the analysis tools that will be integrated in the PATH-SAFE platform for E. coli and L. monocytogenes. Tools for AMR genotyping are discussed separately in Section 3.
Speciator is a tool for assigning a species to an assembled genome and is part of Pathogenwatch (Gladstone et al., 2020). Speciator can accurately assign species for the majority of genome assemblies within seconds. A number of different lineage identification or molecular typing methods for E. coli are in use, including multi-locus sequence typing (MLST) (Public Databases for Molecular Typing and Microbial Genome Diversity: Multi-Locus Sequence Typing, n.d.), core gene MLST (cgMLST) (cgMLST.Org Nomenclature Server (H25), n.d.), HierCC (Zhou et al., 2021) and ClermonTyping (Beghain et al., 2018), and will be implemented in the PATH-SAFE platform. HierCC defines clusters based on cgMLST by calculating distances between genomes using the number of shared cgMLST alleles (Enterobase, n.d.). ClermonTyping is an in silico method that aims to identify E. coli phylotypes proposed by Clermont (Clermont et al., 2000). For L. monocytogenes only MLST and cgMLST are commonly used and will be implemented in the PATH-SAFE platform. For clustering analyses of L. monocytogenes isolates, threshold levels should not be too tight because L. monocytogenes can persist for an extended time in different environments, and genomes of long-term persisting clones can change substantially over time (Fagerlund et al., 2022; Orsi et al., 2021).
The gold standard within-species subtyping for E. coli is phenotypic serotyping with antibodies targeting specific surface somatic (O), flagellar (H) and capsular (K) antigens (Bessonov et al., 2021). Prediction of serotypes from WGS data is possible, for example, by using the ECTyper tool that will be implemented in the PATH-SAFE platform (Bessonov et al., 2021). Other tools for predicting serotypes from WGS data are either out-of-date and no longer maintained, or are still under development and do not yet include E. coli. Prediction of O serotype by ECTyper is reliable, prediction of H serotype is generally reliable but may be wrong for marginal cases. A comprehensive dataset of isolates with known serotypes and sequence data needs to be analysed to identify a specific set of cases for which H serotype prediction is unreliable, but this is out of scope of the current PATH-SAFE release. Similarly, prediction of K serotypes from WGS data is unreliable using currently available tools. Therefore, prediction of K serotypes will not be covered by the current PATH-SAFE release. Although serotyping has historically been the method of choice for subspecies E. coli classification, the advice of the technical community input advisory group (CIAG) is to anticipate a shift towards hierarchical clustering of isolates based on sequence data and design the PATH-SAFE platform accordingly.
Serotype identification is less important in L. monocytogenes than in E. coli because all clinically relevant cases are caused by only three serotypes (1/2a, 1/2b, and 4b) (Borucki & Call, 2003). Therefore, no sequence-based serotyping tools for L. monocytogenes exist or will be implemented for this release of the PATH-SAFE platform.
VirulenceFinder will be integrated in the PATH-SAFE platform to support the identification of virulence determinants from sequence data in E. coli (Kleinheinz et al., 2014). VirulenceFinder provides a more comprehensive annotation of virulence factors than AMRFinderPlus. Adhesins are crucial virulence determinants in E. coli.
Assigning virulence determinants to L. monocytogenes isolates based on sequence data is problematic because of frequently degraded genomes with premature stop codons and frameshift mutations (Nightingale et al., 2008). Therefore, it is difficult to confirm if a potential genetic virulence factor is functional or not. Moreover, gene naming in different virulence factor databases is inconsistent, and different databases seem to include different virulence genes. The Pasteur Virulence Scheme maintained by Institut Pasteur Paris has been selected for inclusion in the PATH-SAFE platform, since it is the only database that includes Listeria-specific genes and is up-to-date (PATH-SAFE, n.d.-a). VirulenceFinder has a Listeria-specific database, but it has not been updated for more than five years. Nevertheless, it may be useful to include VirulenceFinder output in PATH-SAFE reports for comparison.
Identification of plasmids in E.coli was not a high-priority functionality in the survey of PATH-SAFE platform end users and will be integrated into the PATH-SAFE platform in a future development cycle.
Recommendations:
-
As the PATH-SAFE platform is expanded to new food-borne pathogens, integrating a tool to differentiate species will be useful. Speciator has been validated and shown to be 99.9% in agreement with Kraken in correctly assigning E. coli genomes.
-
Tools for MLST and cgMLST should be available on the PATH-SAFE platform for both E. coli and L. monocytogenes. In addition, HierCC and ClermonTyping tools should be available for E. coli.
-
The platform should implement tools for clustering E. coli and L. monocytogenes based on MLST/cgMLST profiles. Clusters should be named according to their HierCC codes.
-
The PATH-SAFE platform should implement a tool for predicting E. coli serotypes from sequence data. ECTyper has been selected as the only up-to-date tool for this purpose. Although serotype determination of E. coli isolates is useful for historical reasons, the platform should be designed in a way that makes it easy to switch to hierarchical clustering of isolates.
-
The identification of genetic virulence determinants is essential in the analysis of E. coli and L. monocytogenes. VirulenceFinder and AdhesiomeR should be implemented in the PATH-SAFE platform for virulence determinant identification in E. coli. The Pasteur L. monocytogenes Scheme has been selected for virulence determinant identification in L. monocytogenes, although none of the available databases seems to include all known genes determining virulence in L. monocytogenes. Therefore, it may be useful to implement VirulenceFinder as a second tool and report both output from the Pasteur scheme and from VirulenceFinder.
3. Genotypic markers of antimicrobial resistance in E. coli and L. monocytogenes
WGS data can be used to predict resistance phenotypes from the presence or absence of known resistance determinants and genetic risk factors but cannot be used to recognise novel resistance determinants. At the same time, genetic AMR markers also serve as important epidemiological markers. AMR is of greater clinical and epidemiological importance in E. coli than in L. monocytogenes. This section will describe drug and risk markers to be monitored for E. coli and L. monocytogenes, which tools for antimicrobial resistance prediction produce the best results for E. coli and L. monocytogenes and specify requirements for the validation of these tools.
3.1. Drugs and risk markers to monitor
Both AMR genes and point mutations linked to resistance to specific drugs or drug classes should be monitored so that platform output can be organised in the style of antibiograms. The PATH-SAFE platform AMR tools will be for risk assessment at the population level only and not for clinical decision-making at the individual level. All resistance markers are potentially useful, regardless of the clinical relevance of the associated drug or phenotype, as they can have epidemiological value in defining and identifying specific outbreak strains and thus aid in communications with partners who do not routinely sequence isolates.
Dictionaries mapping individual markers to specific drugs and drug classes are important for reporting and to organise markers in the form of an antibiogram. There are mainly two databases of risk markers for E. coli and L. monocytogenes, ResFinder and the NCBI Reference Gene Catalog. ResFinder maps genes to drug classes and specific antimicrobials. NCBI Reference Gene Catalog maps genes to drug classes and subclasses, where a subclass is sometimes a drug class and sometimes a specific antimicrobial.
Interoperability with international agencies needs to be considered to avoid confusion due to differences in nomenclature of genes and mutations, or markers that are present in one database and not another. EU surveillance programmes currently utilise the ResFinder database, while US agencies utilise NCBI Reference Gene Catalog.
The ResFinder and the Reference Gene Catalog databases include the same genetic AMR and risk markers with one exception. The L. monocytogenes gene vga(G) is included in the AMRFinderPlus database, but not in ResFinder, and is responsible for the majority of false positive predictions of Clindamycin resistance.
Antibiograms for E. coli and L. monocytogenes are included in Table 2 below:
Recommendations:
-
The platform should include all AMR markers in the source databases, regardless of clinical relevance of the associated drug or phenotype, as they can have epidemiological value in defining and identifying specific outbreak strains.
-
The exception is the L. monocytogenes gene vga(G) which is included in the AMRFinderPlus database but is responsible for the majority of false positive predictions of Clindamycin resistance.
3.2. Tools for antimicrobial resistance prediction from sequence data
As in the previous phase of the PATH-SAFE project focusing on Salmonella, the two tools considered for resistance prediction from sequence data are ResFinder drawing on the ResFinder database and AMRFinderPlus utilising the NCBI Reference Gene Catalog database (Feldgarden et al., 2021; Florensa et al., 2022). ResFinder is used by EU agencies, whereas AMRFinderPlus is used by agencies in the USA. As a result of not including the L. monocytogenes vga(G) gene, ResFinder provides slightly better sequence-based AMR predictions for L. monocytogenes than AMRFinderPlus. Otherwise, both tools are largely consistent in their predictions, and both produce non-negligible amounts of false positive and false negative predictions. For interoperability with international agencies involved in food security and food-borne disease surveillance, the PATH-SAFE platform should facilitate comparability with both ResFinder and AMRFinderPlus output and offer both tools to users.
Recommendations:
-
For simplicity of software engineering, the platform should implement existing tools rather than creating another marker database and drug dictionary that need to be tested and maintained.
-
For interoperability with international agencies, the platform should facilitate comparability with both ResFinder and AMRFinderPlus output.
-
When predicting AMR from sequence data in L. monocytogenes, predictions made by AMRFinderPlus based on presence of the L. monocytogenes vga(G) gene need to be interpreted with caution because this gene is responsible for the majority of false positive predictions of Clindamycin resistance.
3.3. Validation of antimicrobial resistance prediction tools
Validation of genomic AMR prediction tools is essential for their use in FBD surveillance. There are three ways to conduct a validation, 1) using an alternative genomic analysis method than those used by the prediction tools, 2) conduct PCR analysis for key resistance markers, or 3) compare genotypic resistance predictions to phenotypic resistance profiles. Validation of tools integrated into the PATH-SAFE platform employed method 1, comparison of alternative AMR prediction tools. The comparison focused on ResFinder and AMRFinderPlus. Comparison of genotypic resistance predictions to phenotypic resistance is out of scope of the PATH-SAFE project. The validation reports can be found in Appendix 1.
Recommendations:
-
Validation should be done for isolates from all sectors involved in food security, as markers may be distributed differently.
-
Comparison to phenotypes is not within the scope of this platform which is geared towards cross-sector surveillance. It is envisaged that agencies will conduct phenotyping in addition to sequencing where this is considered important. Validation could be done by stakeholders against their own current phenotyping methods, if considered important for them.
-
Validation against alternative genomic analysis methods should focus on comparison to ResFinder and AMRFinderPlus. Since both tools will be implemented in the platform, this validation is essentially built-in.
-
Comparison to alternative genomic analysis methods should focus on comparison to outputs of existing pipelines currently utilised within the stakeholder agencies, so that differences can be understood and potentially addressed (as these approaches are likely to be run in parallel there is potential for confusion).
4. Data standards for genomic surveillance of E. coli and L. monocytogenes
Data standards, including metadata and quality control (QC) standards and reporting criteria, are critical to enable comparability and meaningful joint analysis of datasets shared by different agencies on a shared platform. Recommendations on data standards need to be implemented in a way that does not disrupt existing analysis pipelines.
4.1. Metadata requirements
The metadata framework that the PATH-SAFE project has developed for Salmonella has been adapted for the species E. coli and L. monocytogenes. Mandatory metadata fields required for submission to the platform are intentionally kept minimal to comply with UK GDPR requirements, follow the principle of data minimisation, and to avoid accidental leaking of sensitive data. Since the PATH-SAFE platform will be only accessible to designated personnel of participating agencies, these agencies will be able to share additional metadata that could not be shared on a research-focused platform.
Recommendations:
-
The number of metadata fields should be small initially to facilitate upload of data and use of the PATH-SAFE platform and to be consistent with UK GDPR obligations.
-
Minimum metadata requirements of the platform should be compatible with the metadata collected by each agency. They should also be compatible with concerns around data sharing and legal obligations.
-
All metadata must be processed in line with UK GDPR guidelines and align with organisational policies and relevant legislation.
-
The PATH-SAFE platform should implement a gated access model that will allow participating agencies to share additional metadata with trusted partners and at the same time minimise the risk of leaking sensitive information.
4.2. Quality control requirements for metadata
All genomes assembled by the PATH-SAFE platform’s assembly pipeline will be added to the database but hidden if they fail read or assembly QC thresholds. The same QC metrics that are used for Salmonella genomes will also be used for E. coli and L. monocytogenes genomes (Table 2). However, the threshold values will depend on the pathogen species. As no canonical QC metrics for E. coli and L. monocytogenes have been defined, mean and standard deviation values were calculated for genome size and GC content from representative genomes in the RefSeq database (PATH-SAFE, 2025a, 2025b). If this systematic and mathematical way of defining QC metrics is accepted as a standard way of calculating QC metrics, it can also be applied to new organisms. Other methods for calculating threshold values are possible. For example, a recent paper evaluating data from the network of the European Union Reference Laboratory for Antimicrobial Resistance genomic proficiency tests set QC thresholds three standard deviations from the mode (Sørensen et al., 2024).
E. coli harbours large plasmids with high AT content (up to 40%) which could reduce the measured GC content of the E. coli genome (Silva et al., 2020). Therefore, a lower threshold of 37.5 % has been selected for GC content as QC metric for E. coli sequences (Table 3). Moreover, RefSeq mainly contains human isolates. However, E. coli isolates from animal hosts are known to be more diverse than those from human hosts (Lagerstrom et al., 2024). Consequently, QC metrics may need to be updated if E. coli sequences from animal isolates tend to fall outside of the range defined by QC metrics determined from RefSeq genomes. Due to the large genetic variability found in E. coli, QC threshold values need to be relatively more permissive so as not to erroneously exclude sequence data.
In order to avoid errors introduced by manual upload of sequence data and to reduce the workload on agency staff, the PATH-SAFE platform should have functionality for automated data upload on a regular basis (for example, daily checks for new data followed by upload of new data). The automated data upload should be implemented to be compatible with firewalls that protect individual agencies’ data systems. For example, the CLIMB-COVID platform stored data in S3 buckets with a secure https endpoint that is accessible from behind most firewalls (Nicholls et al., 2021). Additional work may be needed to ensure secure automated upload is possible from within each participating agency’s data system.
Recommendations:
-
The PATH-SAFE platform should have an automated QC mechanism for validating uploaded metadata.
-
For ease of use, options for both bulk upload of metadata and for upload of individual metadata fields should be offered by the platform. In addition, functionality for regular automated uploads could be provided.
-
The QC criteria in Table 3 should be used for the PATH-SAFE platform.
5. International best practices and reporting standards in genomic surveillance of E. coli and L. monocytogenes in food-borne disease
This section recounts international case studies meant to illustrate best practices for genomic surveillance of E. coli and L. monocytogenes. Key lessons learnt are summarised after each case study.
5.1. Impact of WGS in routine public health surveillance of Listeria – a decade of genomic surveillance in the UK
The UK Health Security Agency (UKHSA) is responsible for protecting every member of every community from the impact of infectious diseases. The UK surveillance system employs a three-pronged approach combining microbiology, epidemiology and bioinformatics. Within UKHSA the Gastrointestinal Bacteria Reference Unit (GBRU) is the national reference laboratory for gastrointestinal pathogens and provides specialist testing of clinical, food and environmental samples (UK Health Security Agency, 2024). GBRU’s activities include bacterial typing for national surveillance, detection and investigation of outbreaks, and specialist microbiological services for the examination of clinical, food, water and environmental samples. GBRU relies on a combination of phenotypic and genotypic methods, including WGS, to detect, identify and type gastrointestinal and food-borne bacterial isolates. GBRU works closely together with the Gastrointestinal Infections and Food Safety One Health (GIFSOH) Division. GIFSOH is the national centre for surveillance of gastrointestinal pathogens and other food and environment transmitted infections in England and Wales and is responsible for identifying and managing national outbreaks of gastrointestinal infections.
National enhanced surveillance for listeriosis has been undertaken in England and Wales since 1992. Case ascertainment is by mandatory reporting under the Health Protection Notification Regulations (2010) of laboratory-diagnosed cases from clinical microbiology laboratories via an electronic system. GBRU started transitioning to WGS surveillance of L. monocytogenes in 2010. As part of case reporting and outbreak detection, hospital staff collect clinical details, and a food preferences questionnaire is administered to patients of family members. The questionnaire captures food exposures up to three weeks before onset of illness. L. monocytogenes cultures from clinical cases are submitted to GBRU on a voluntary basis.
According to GBRU’s estimates, there are on average 183 listeriosis cases in the UK per year, including those reported to national surveillance and unreported cases in the community (O’Brien et al., 2016). In 2023, GBRU sequenced 1118 L. monocytogenes isolates and 9172 in total since 2015.
The advantages of using WGS for surveillance are:
-
Short turnaround times (6-10 days from culture to results, depending on duration of culture growth and timing of sequencing services)
-
Reduced analysis costs per specimen
-
High-quality data
-
Standardisation and simplified global comparison between strains
-
Safety in laboratories (compared to handling live specimens)
-
Improved resolution for strain discrimination
-
Ready inclusion of phylogenetic analysis of WGS data
-
Improved detection and surveillance of outbreak clusters
-
Rapid screening for virulence and AMR markers
-
Overall improved understanding of pathogen evolution and pathogenicity
WGS data from gastrointestinal pathogens may enter the UK disease surveillance system via two alternative routes, the hospital and the laboratory route. In the hospital route, pathogens are isolated from patient samples in the hospital laboratory and sent for WGS typing to the national reference laboratory which may lead to detection of an outbreak. In the laboratory route, the national surveillance system is notified of a case which leads to patient follow-up and public health action.
Once WGS data has entered the surveillance system, it is submitted to SnapperDB for storage and analysis, including SNP distance calculation and clustering (Dallman et al., 2018). WGS data is stored in the Gastro Data Warehouse (GDW), UKHSA’s internal database system for WGS surveillance of gastrointestinal pathogens (UK Health Security Agency, 2013). GDW stores the following metadata fields:
-
Next Generation Sequencing (NGS) ID
-
Specimen source (e.g. human, animal)
-
Receipt Date
-
Report Date
-
Country (e.g. England, Wales)
-
Travel history (less important for L. monocytogenes, more important, for example, for E. coli or Salmonella)
-
Travel destination
-
Organism identified
-
Sequence type (ST)
-
Lineage
-
SNP address
-
Phylogeny
Data submitted to the GDW is analysed with an automated algorithm that detects clusters of two or more isolates within a distance of five SNPs. Subsequently, a prioritisation algorithm analyses WGS data in GDW together with epidemiological data collected by GIFSOH to detect outbreaks and, if necessary, to trigger a defined response structure managed by UKHSA working with partners in respective lead roles. Prioritisation takes the following factors into account:
-
Cluster characteristics (size, growth rate, case demographic characteristics)
-
Severity of infection and invasiveness index
-
Within-cluster diversity and phylogenetic context
-
Genotypic AMR profile
-
Epidemiological and ecological context
-
Related food/animal/environmental isolates
-
‘X’ factors (e.g. special cluster epidemiology or phylogeny, probability of public health action and legal basis)
-
Resource availability
UKHSA implements an open data sharing policy for food-borne pathogen genomes via automated overnight uploads to the SRA. As a result, the UK is the second biggest contributor of food-borne pathogen genomes to international sequence databases after the US. For example, 10% of all L. monocytogenes genomes in the NCBI Pathogen Detection Database have been uploaded by UK agencies.
From 2015-2021, the period of WGS implementation for L. monocytogenes surveillance, 83 clusters were identified, and the source of infection was identified 13 times (two times per year) doubling the number of source identifications compared to the pre-WGS era (1981-2015, 28 time source identified, corresponding to one source identification per year). In particular, a matching food isolate was detected before the first case isolate for 56% of clusters. This means that there is a window of opportunity in which public health action can be taken to prevent or curb outbreaks early.
An example of the advantages of WGS for L. monocytogenes surveillance is the frozen vegetable outbreak from 2016-2021. In March 2022, cooked dried carrots from a producer of frozen vegetables were found to be contaminated with L. monocytogenes clonal complex 20 (CC20). An investigation was started looking for sequences of similar isolates in the GDW less than 5 SNP differences apart. A clinical isolate from 2021 was identified, as well as isolates from potatoes, swede, parsnips and sweet potatoes from the same producer from February 2022 belonging to the same CC20 and within 10-25 SNPs distance. A subsequent search for all sequenced isolates belonging to a cluster within 25 SNPs found seven additional cases, including two deaths, that occurred in England, Northern Ireland and Scotland over a timeframe from January 2016 to December 2021. Moreover, sequence data showed that isolates of the outbreak cluster were resistant to Benzalkonium (emrC gene) and quaternary ammonium compounds (qacC gene).
Key lessons learnt:
-
WGS has enhanced food-borne pathogen surveillance at an unprecedented resolution. This has allowed the identification of clusters of L. monocytogenes which was not possible in the pre-WGS era and doubled the success rate for source identification.
-
WGS for surveillance of L. monocytogenes has accelerated turnaround times from sample isolation to results.
-
Collaboration of reference laboratories that carry out sequencing analyses and epidemiological and One Health units that provide metadata is critical for prioritising isolates for outbreak investigations.
-
A big advantage of WGS data is that it is stored electronically by nature and can be analysed by automated algorithms speeding up time to results and saving staff resources.
-
As L. monocytogenes can persist in the environment for extended periods and undergo evolution, flexible threshold values should be employed for clustering for outbreak detection and source identification.
-
Further progress could be made by developing and implementing new technologies and methods to understand accessory genomes (prophages, plasmids) that can carry AMR determinants. Moreover, genomic markers within L. monocytogenes strains for routine virulence assessment, clinical severity prediction, resistance to cleaning products should be developed.
5.2. The Swiss Pathogen Surveillance Platform - a secure online platform for One Health pathogen surveillance
The Swiss Pathogen Surveillance Platform (SPSP) is a secure online platform for One Health pathogen surveillance. SPSP is a project by the University of Zurich, the University of Lausanne and the University of Geneva and is hosted by the Swiss Institute of Bioinformatics (Neves et al., 2023). Its aim is to generate high-resolution data for One Health multidrug-resistant (MDR) bacterial infections and source attribution. To increase trust in the platform, it has been ISO accredited.
SPSP is linked to other databases, including the Swiss Centre for Antibiotic Resistance (ANRESIS) and European genomic surveillance platforms by EFSA and ECDC. Ridom is installed on the cluster to facilitate data exchange with German and Austrian reference laboratories (Ridom Bioinformatics, n.d.). Moreover, SPSP is linked to Agroscope, the Swiss centre of excellence in agricultural research, for sharing of food isolates (Schweizerische Eidgenossenschaft, n.d.), and to NENT, the Swiss National Centre for Enteropathogenic Bacteria and Listeria that is responsible for the analysis of human isolates (Universität Zürich, n.d.). Food producers are currently not obliged to share sequence data with public health authorities, but they will have to do so from 2027 as per changes in the relevant legislation.
SPSP started with methicillin-resistant Staphylococcus aureus MRSA as an exemplar pathogen. A bottom-up approach was implemented through a pilot starting with the Swiss Institute of Bioinformatics (SIB) and two hospitals. Once the pilot had successfully demonstrated the platform’s capabilities, new partners joined.
SPSP proved its utility and functionality during the COVID-19 pandemic during which the Swiss Federal Office of Public Health (FOPH) received three automated reports per week from the platform. SPSP also shares SARS-CoV-2 sequence data with GISAID. Sensitive metadata are not shared outside of SPSP. The median time from a positive PCR test to sharing data with GISAID was 14 days. A policy that sequencing was only reimbursed by the government if it happened fast enough promoted fast turn-around times. Now SPSP is used for many pathogens and is partially funded by the FOPH.
A key achievement of SPSP is a transparent governance structure to build trust among users submitting sequence data to the platform. Universities sequenced pathogens even before the COVID-19 pandemic but withheld their data until analyses of the data were published in a journal. SPSP’s governance ensures that data generators retain rights to the data and are acknowledged in publications using the data.
Switzerland incentivises data sharing in the health system via the Swiss Personalised Health Network (SPHN) which closely collaborates with SIB. BioMedIt provides the backbone of the SPHN data system. Hospitals can log into a secure node and upload data protected by a firewall so that even sensitive information can be shared. New users of the system first need to pass an exam before they are granted access. It takes a week until a new partner organisation is enrolled and online on the platform. SPSP utilises the BioMedIt backbone. This ensures that, although SPSP has been built as a research platform, FOPH can also use the data on the platform for surveillance.
The stringent security of SPSP has three layers: 1) only whitelisted IPs can access the platform, 2) users need to log into the SIB access zone, 3) SIB sensitive zones ensure that only authorised users can access certain sensitive data. However, the platform sends automated notifications to users if a similar isolate is uploaded.
The legal framework underlying the trusted governance structure took years to develop. The consortium agreement prescribing SPSP’s governance was signed in January 2011. Governance structures include an executive board, a scientific board and an advisory board. Since Swiss law allows data sharing with FOHP for surveillance during outbreaks but not for research, SPSP’s governance framework provides prospective data sharing agreements to facilitate timely data transfer among collaborating platform users.
Standardised metadata fields are available following recommendations of the Swiss Biobank which is aligned with the European Biobank (BBMRI-ERIC). For example, species identification follows the NCBI taxonomy. FOPH may even use residence address data of reported cases for public health action. Researchers do not have access to address data but may have access to geographic data with which spatial modelling can be carried out to inform public health decisions.
SPSP’s bioinformatics pipeline is modular and based on Nextflow (Nextflow, n.d.). New tools that are considered for integration into SPSP are discussed regularly by the Scientific Advisory Board. If the decision is made to integrate a new tool, it is first tested for a period of time. Tools are selected or deselected by direct voting of Scientific Advisory Board members. FOPH is updated on new tools and scientific developments that are of interest for public health and surveillance at least once a year.
Next planned steps for the development of SPSP include:
-
Expansion of the frontend for new bacterial pathogens and queries
-
New dashboard with visualisation of phylogenies
-
Integration of Nextsrain (Nextstrain, n.d.)
-
Bacterial pathogen-specific pipelines, for example, integration of Kleborate (Lam et al., 2021)
-
Automatic flagging of clusters
Key lessons learnt:
-
ISO accreditation improves trust in platforms for genomic surveillance of food-borne and One Health pathogens.
-
Exchanging data with national and international partner agencies enhances FBD surveillance and needs interoperable interfaces for information exchange. This includes standardised metadata fields.
-
Building a genomic surveillance platform from the bottom-up, including a pilot phase with a few selected partners, can demonstrate the functionality and benefits of the platform and improve acceptance among new partners.
-
Stringent security and multi-tier access control is required to maintain trust in the platform. Nevertheless, it is possible for the platform to share relevant information among partners. For example, if similar isolates are uploaded by different users of the platform, both users receive automated notifications which can trigger further investigations. This would not be possible or only with a delay in the absence of a common surveillance platform.
-
Timely data transfer among collaborating platform users can be facilitated by including prospective data sharing agreements in the platform’s governance and legal frameworks.
-
New analytical tools that may be useful for public health surveillance are tested for a period of time before they become an integral part of the platform.
-
A platform for different user groups needs to balance the interest of the different user groups by appropriate governance bodies and a tailored communications strategy to inform different user groups of updates to the platform.
5.3. Genomic-driven PulseNet Canada for FBD surveillance in Canada
In Canada, each year one in eight people, corresponding to more than 4 million Canadians, fall ill from contaminated food. Each year, 11,500 hospitalisations and 240 deaths occur as a consequence of FBD (Thomas et al., 2013). Bacterial pathogens are responsible for 66% of hospitalisations and 76% of deaths due to FBD (Thomas et al., 2013). Listeria is the cause of 4% of hospitalisations and 33% of deaths. STEC is responsible for 11% of hospitalisations and 12% of deaths. For comparison, Salmonella is responsible for 25% of hospitalisations and 16% of deaths.
PulseNet Canada is a network of Provincial Public Health and Federal laboratories that upload WGS data to a central database for national surveillance, outbreak detection and response. At the federal level PulseNetCanada partners with four interconnected programmes. The National Enteric Surveillance Program (NESP) is responsible for timely reporting and analysis of enteric disease cases, including, for example, weekly reports of serotype prevalences. NESP data is private but can be shared upon request. Summary data are published in annual reports. FoodNet Canada performs detailed integrated surveillance at four sentinel sites or regions, including the collection of retail food samples, farm samples, surface water samples and enhanced data collection of clinical cases in the sentinel regions. An interactive summary of FoodNet Canada data is publicly available (Government of Canada, 2024). The Enhanced National Listeriosis Surveillance Program (ENLSP) collects detailed epidemiological information on all listeriosis cases. The Canadian Integrated Program for AMR Surveillance (CIPARS) collects and analyses trends in antimicrobial use and resistance. PulseNet Canada works closely with federal epidemiologists at the Centre for Foodborne, Environmental and Zoonotic Infectious Diseases (CFEZID) to detect and respond to foodborne disease threats. Agencies collaborate according to the protocols laid out in Canada’s Foodborne Illness Outbreak and Response Protocol (FIORP) (Public Health Agency of Canada, 2017).
Genomic surveillance of FBD in Canada begins with Provincial Public Health Laboratories collecting isolates and data from clinical cases. Provincial laboratories, the Canadian Food Inspection Agency, FoodNet Canada and others collect non-clinical isolates including food, animal and environmental isolates. Priority pathogens for national surveillance are Salmonella, STEC, Listeria, Shigella, Vibrio cholerae, Vibrio parahaemolyticus, Campylobacter, Yersinia, Cronobacter, Cryptosporidium, and Cyclospora.
PulseNet Canada was established in 2001 to harmonize FBD surveillance systems across Canada and with international partners including PulseNet USA. Analyses were first based on pulsed-field gel electrophoresis (PFGE) (Public Health Agency of Canada, 2022). By 2017 WGS had been validated by PulseNet Canada and replaced PFGE as the primary molecular analysis tool. WGS surveillance for FBD started with Listeria and Salmonella in 2017 and E. coli and Shigella in 2018. Initially, WGS and PFGE were used in parallel. PulseNet Canada employs a decentralised data model. Partner laboratories and the National Microbiology Laboratory generate WGS data and upload them to IRIDA for isolate level analyses such as in-silico serotyping. In silico serotype prediction for Salmonella is done with SISTR and for E. coli ECTyper is used (Bessonov et al., 2021; Yoshida et al., 2016).
Nearly 4,000 whole-genome sequences from non-clinical (food, environmental) isolates and more than 10,000 whole-genome sequences from clinical isolates are processed by PulseNet Canada annually. The sequences are from Salmonella, Listeria, STEC, Shigella, and V. cholerae. WGS surveillance for Campylobacter, Cronobacter, Vibrio parahaemolyticus, and Yersinia enterocolitica are the next priorities to undergo validation.
Data is transferred from the national IRIDA database to pathogen-specific Bionumerics-databases which are used for aggregate analysis and surveillance reporting.. However, BioNumerics was discontinued at the end of 2024 (Wikipedia, 2023). To replace the functionality of Bionumerics, Canada’s National Microbiology Laboratory and PulseNet Canada have been developing their own Genomic Surveillance Platform (Public Health Agency of Canada, 2022). As part of this replacement the toolchain for WGS surveillance of FBD is undergoing a general overhaul. Table 4 shows currently used tools and tools planned to be integrated into the workflow going forward
The Canadian Network for Public Health Intelligence (CNPHI) platform is used for communicating surveillance results and for planning public health interventions. Jurisdictions across Canada are represented on CNPHI which has more than 7100 users (Public Health Agency of Canada, 2023).The CNPHI platform offers functionality to support surveillance and laboratory work, public health alerts, knowledge management and early warning, and collaboration and response (Public Health Agency of Canada, 2023). In short, IRIDA Next is used for pulling together data and CNPHI is used by public health staff to share insights.
A major success story for WGS-based FBD surveillance in Canada is the detection of and response to 16 multi-jurisdictional outbreaks of Salmonella associated with frozen raw breaded chicken between 2017 and 2019 (Kerr et al., 2024). The outbreaks comprised 487 human cases ranging in age from 0 to 98 years and resulting in 79 hospitalizations and two deaths. As a consequence of WGS-based outbreak detection, 14 frozen raw breaded chicken products were recalled, and one was voluntarily withdrawn from the market (Kerr et al., 2024). The detection of repeated outbreaks associated with raw frozen chicken also led to the issuance by the Canadian Food Inspection Agency (CFIA) of new industry requirements at the level of the manufacturing plant to reduce contamination with Salmonella. After the industry deadline for implementing the new requirements in 2019, no new Salmonella outbreaks associated with frozen raw breaded chicken were observed (Kanoatova et al., 2024). Data collected as part of an intervention study with a comparison group methodology showed that Salmonella prevalence decreased from 28% before 1 April 2019 to 2.9% after the implementation of the new CFIA requirements (Kanoatova et al., 2024). Overall, salmonellosis incidence was reduced by 23% (Glass-Kaastra et al., 2022). It has been estimated that the new CFIA requirements reduced salmonellosis cases in 2019 by 26,000 compared to the previous five years. It has also been estimated that the measures led to 213 fewer hospitalisations and two fewer deaths [ref]. The reduction in salmonellosis-associated illness and mortality led to an estimated CA$ 20 million, including CA$ 13 million saved in terms of premature mortality, CA$ 5 million in lost productivity and CA$ 2 million in reduced medical costs (Glass-Kaastra et al., 2022).
Key lessons learnt:
-
WGS-based surveillance has enabled improved outbreak detection at greater temporal and spatial resolution.
-
The additional information gained from WGS can support the issuance of new regulatory directives to reduce FBD burden.
-
WGS-based surveillance has contributed to reducing FBD burden and achieved major cost reductions associated with illness, mortality and lost productivity.
-
Successful public health surveillance does not only rely on efficient tools for aggregating and analysing data but also on effective communication tools.
6. Conclusion
WGS surveillance is expected to increasingly inform interventions for food safety and public and veterinary health, including for E. coli and L. monocytogenes.
This technical note summarises recommendations made by Community Input Advisory Groups on the design and capabilities of the PATH-SAFE platform and on the application, technical requirements and best practices for WGS in E. coli and L. monocytogenes surveillance.
Documentation of the PATH-SAFE platform and validation reports are provided in separate documents.
7. Biographies of Community Input Advisory Group Members
Dr Manal AbuOun is a scientist at the Department for Environment, Food and Rural Affairs (DEFRA) in the Department of Bacteriology and Food Safety.
Dr. Marc W. Allard is a Senior Biomedical Research and Biomedical Product Assessment Services Officer in the Division of Microbiology in FDA’s Office of Regulatory Science. Dr. Allard joined The US FDA in 2008 where he uses Whole Genome Sequencing (WGS) of foodborne pathogens to identify and characterise outbreaks of bacterial strains, particularly Salmonella, E. coli, and Listeria. Dr. Allard specialises in both phylogenetic analyses, as well as the biochemical laboratory methods which generate the WGS information. Dr. Allard helped develop the first distributed network of laboratories that utilize whole genome sequencing for pathogen identification and traceback called the GenomeTrakr database, which is part of the NCBI Pathogen Detection web site. These tools are used daily for outbreak investigations and compliance. Dr. Allard acts as senior scientist to advise the US FDA on both WGS and phylogenetic methods as they apply to public health.
Professor Matthew Avison leads a research group studying AMR in bacteria. His research group uses molecular genetics, biochemistry and functional genomics techniques to identify and characterise AMR mechanisms in key human pathogens, their mobilisation, and their control and then uses this information to combat the problem of AMR by developing interdisciplinary research collaborations. He leads the One Health Selection and Transmission of AMR (OH-STAR) consortium, the One Health Drivers of Antibacterial Resistance in Thailand (OH-DART) consortium, and contributes to the FARMS-SAFE consortium to survey AMR and antimicrobial usage and to identify the drivers of AMR in Argentinian Pig and Dairy farming systems.
Dr Kate Baker is Principal Investigator at the Department of Genetics at the University of Cambridge. She studies how pathogen genome variation and evolutionary processes impact their epidemiology and control. She has a particular interest in the dynamics of the accessory genome in bacterial populations, including antimicrobial resistance. Using a combination of microbial genomics, epidemiological approaches, and molecular microbiology, her research group unpicks disease processes at both patient and public health levels in both high-income and lower- to middle-income nation settings in collaboration with clinicians, public health practitioners, in vivo experimentalists, and mathematical modellers. She also has an interest in knowledge exchange and policy and has held various external secondments (GO-Science, SEDRIC, and UKHSA).
Antonia Chalka is a doctoral candidate at the University of Edinburgh. Her current project uses machine learning to investigate Salmonella host specificity and is under the supervision of Professors David Gally and Mark Stevens.
Professor Tom Connor is the Head of the Public Health Genomics Programme within Public Health Wales, the national Public Health Institute of Wales, and is a Professor of Bioinformatics and Pathogen Genomics at Cardiff University. He earned his PhD from Imperial College, before undertaking a PostDoc at the Wellcome Sanger Institute. Following this, in 2012, he took up his permanent academic position at Cardiff University. He has a research track record that has seen his team apply genomics to examine microbial populations, to understand their evolution and, in the case of pathogens, their genomic epidemiology. As an extension to his research work, Tom has provided leadership as part of the development of national computational infrastructures - such as the MRC CLoud Infrastructure for Microbial Bioinformatics (CLIMB) and Supercomputing Wales - which are designed to support genomics and bioinformatics research. Since 2016 Tom has worked to translate his research expertise into clinical service, as part of his role within Public Health Wales. Within Public Health Wales, Tom has led the development of bioinformatics as an area of activity, as well as developing the pipelines and infrastructure that underpin current clinical services. As part of this work Tom has overseen the development of ISO 15189 accredited clinical genomics services including for C. difficile (first service of its type in the world), HIV (first national service of its type in the UK), SARS-CoV-2 (first accredited SARS-CoV-2 genomics service in a UK public health agency). Collectively these services have sequenced almost 300,000 pathogen samples from Welsh patients to support services ranging from individual patient management to national-scale pandemic monitoring and response.
Professor Alistair Darby is Co-Director of the Centre for Genomic Research (CGR) at Liverpool and has more than 15 years experience as an applied entomologist. His main area of research is in microbial/host interactions which covers his interest in arthropod symbiosis, pathogens and microbiomes. He has extensive experience in designing and analysing genomic and RNAseq data, including arthropod genomes; e.g., Nasonia wasp (Science), tsetse fly (Science), mite (GigaScience), and a new high quality version of the Diamondback moth genome sequence using the PacBio (data at lepbase.org). His research also uses single cell genomics and he is the lead for the CGR single cell genomics facility.
Dr Daniel Dorey-Robinson After completing his PhD at the APHA/Uni of Surrey he spent 7 years working in bioinformatics looking at veterinary virology, bacteriology and immunology, before joining the Defence science and technology laboratories in 2022 where he is a senior computational biologist.
Professor Adrian Egli is the Director of the Institute of Medical Microbiology at the University of Zurich. With a career rooted in a profound understanding of host-pathogen interactions, his research leverages cutting-edge technologies, including genome sequencing, mass spectrometry, and advanced data analysis through bioinformatics and machine learning. He aims to revolutionize the diagnosis, treatment, and prevention of infectious diseases, underscoring his commitment to advancing medical science and healthcare outcomes. He is co-founder of the Swiss Pathogen Surveillance Platform.
Dr Richard Ellis is Head of Genome Analysis at the Animal and Plant Health Agency. After completing his DPhil from Oxford, he spent over 10 years in academia studying bacterial ecology and population genetics before joining APHA. His work now focuses on the application of high-throughput sequencing technologies for understanding the evolution and spread of pathogens, for investigating diseases of unknown aetiology, and the integration into disease surveillance programmes. He oversees the laboratory teams responsible for routine disease testing required for both surveillance and trade, and the capacity to scale up testing in the event of a disease outbreak.
Professor David Gally holds a personal chair in Microbial Genetics at the University of Edinburgh and has been part of the Roslin Institute since 2011. His background is in microbiology and food safety. He was seconded to Food Standards Scotland as their Chief Scientific Advisor from 2021-24. David has led three successive Roslin Institute Strategic Programme grants funded by the BBSRC, the latest on the ‘Control of Infectious Diseases’ in Livestock (2017-2022). His group has been applying machine learning to source attribution for bacterial isolates associated with human infections through food or water, in particular Salmonella and E. coli, to help with outbreak investigations and ongoing work has moved into predictive phage therapy.
Dr Matthew Gilmour leads the ‘Listeria and other Invasive Pathogens’ research group at the Quadram Institute in Norwich, England. Matthew is also deputy lead of Quadram’s ‘Microbes and Food Safety’ strategic programme which has a focus on translating the Institute’s key microbiology findings and genomic technologies with partners in the food industry and government.Matthew was previously based in Canada where his group was a pioneer in using bacterial genomics to study outbreaks, including the large Canadian listeriosis outbreak in 2008 and then the Haitian cholera outbreak of 2010. With this experience in public health, from 2015 to 2020 Matthew was the Scientific Director General of Canada’s National Microbiology Laboratory. In the UK, Matthew is also now Director of the Food Safety Research Network, based at the Quadram Institute. This network has the goal of brokering collaborative research projects between food businesses and academic research groups that will make UK foods safer from microbial risks.
Professor David Graham is Professor of Ecosystems Engineering at Newcastle University who has led AMR surveillance and related projects around the world for over 30 years. He is a member of the United Nations (UN) Quadripartite Technical Group on Integrated Surveillance on Antimicrobial Use and Resistance and is the integration lead on the new Technical Guide on cross-sectoral AMR surveillance for member states. Graham was co-lead author on the UN report, “Bracing for Superbugs: Strengthening environmental action in the One Health response to antimicrobial resistance” and co-wrote the World Health Organisation recommendations aimed at improving water quality, sanitation, and hygiene (WASH) as a strategy for reducing AMR in the low-resource settings. Domestically, Graham worked on UK SAGE sub-group on COVID-19 Transmission in the Wider Environment and was in the Expert Advisory Group who operationalized UK wastewater surveillance during the COVID-19 pandemic. He is a Chartered Engineer, but uses microbiology, genetics, and genomics to create holistic solutions. His current work focuses on relationships between human behaviour, water security, AMR, and sustainable development.
Dr Edward Haynes is the PATH-SAFE Science Fellow. He is a molecular biologist by training, with a background in bacterial genomics of foodborne, plant and honeybee pathogens. His current role is shared between his molecular biology responsibilities at Fera Science and PATH-SAFE Science Fellow. Earlier in his career, he held another joint post between Fera and the FSA to develop genomic approaches to foodborne bacteria, which involved working with the US Food and Drug Administration (FDA) to learn from the extensive work they had been doing to routinely apply genomic tools to foodborne pathogens in the GenomeTrakr project. In the last few years, he has also led projects applying non-targeted metagenomic sequencing approaches to detect AMR genes in different parts of the food production chain, in meat factories and in ready-to-eat foods.
Professor Rene S. Hendriksen is a professor at the Technical University of Denmark, National Food Institute, and leads the Research Group of Global Capacity Building. He is the director of the World Health Organization (WHO) Collaborating Centre, Food and Agriculture Organization (FAO) Reference Center, and European Union Reference Laboratory forFood, Feed and Animal Health as well as Public Health in the area of Antimicrobial Resistance (AMR) and Genomics (WHO), respectively. In his early days, the focus was on conventional and molecular microbiology. Since 2010, he has embraced the era of genomics with a strong focus improving global surveillance of antimicrobial resistance through strengthening activities and implementing and applied research primarily in EU but also Southeast Asia and sub-saharan Africa.
Professor Jay Hinton did his first degree in Microbiology, his PhD at the University of Warwick and his postdoc at the University of Oxford’s Weatherall Institute of Molecular Medicine. Jay became Head of Molecular Microbiology at the Institute of Food Research in Norwich in 1999, and relocated to Trinity College Dublin in 2009. Jay moved his research group to the University of Liverpool in 2012, where he is currently the Professor of Microbial Pathogenesis. In 2003, Jay’s group pioneered a transcriptomic approach that revealed a “snapshot” of Salmonella gene expression during the process of infection of mammalian cells. They discovered a key mechanism for silencing gene expression in bacteria in 2006 mediated by the H-NS protein. At Trinity College Dublin, his team employed RNA-seq-based approaches to understand the transcriptome of Salmonella Typhimurium which involved the identification of 280 non-coding sRNAs. At the University of Liverpool, Jay’s research group is using functional genomics to understand how new Salmonella pathovariants are causing endemic bloodstream infections across sub-Saharan Africa. This disease, iNTS, has killed around 500,000 people over the past decade. His team is now using a combination of molecular microbiology, genomics and functional transcriptomics to generate new insights. Jay led the 10,000 Salmonella genomes project (https://github.com/apredeus/10k_genomes), which sequenced bloodstream isolates from about 50 countries dating from 1949 to 2017, with ~80% representing African and Latin-American datasets. Recent discoveries include the determination of the role of a single noncoding nucleotide in the over-expression of a key virulence factor in African Salmonella (Hammarlöf et al, PNAS) and an understanding of the step-wise evolution of African Salmonella (Pulford et al, Nature Microbiology).
Professor Kat Holt is a computational biologist specialising in infectious disease genomics. She is Professor of Microbial Systems Genomics at the London School of Hygiene and Tropical Medicine (LSHTM) Department of Infection Biology, and Co-Director of the LSHTM AMR Centre. She is also an Adjunct Professor (Research) at Monash University’s Department of Infectious Diseases in Melbourne, Australia; a HHMI-Gates International Research Scholar; and former Editor-in-Chief of the UK Microbiology Society journal Microbial Genomics. Kat has a BA/BSc from the University of Western Australia majoring in Biochemistry, Applied Statistics and Philosophy, with Honours in Genetics (2004); a PhD in Molecular Biology from the University of Cambridge and the Wellcome Trust Sanger Institute on the genomics of typhoid fever; and a Masters in Epidemiology from the University of Melbourne (2011). She has held Early Career (2010-2013) and Career Development (2014-2017) Fellowships from the NHMRC of Australia, and a Senior Medical Research Fellowship from the Viertel Foundation of Australia (2018-2021). Kat and her research team are particularly interested in the global health crisis of AMR, using genomic epidemiology tools to understand the evolutionary history and global dissemination of multidrug resistant pathogens, and developing new tools for prospective surveillance and tracking of emerging problems in the public health and clinical infectious disease space. Kat is a founder and coordinator of the Global Typhoid Genomics Consortium and the KlebNET Genomic Surveillance Platform.
Dr Claire Jenkins started working for UKHSA, formerly PHE, as a Clinical Scientist in 1996. She became head of the E. coli Reference Laboratory in 2012 and deputy head of the Gastrointestinal Bacteria Reference Unit in 2014, the same year as WGS was implemented as the routine typing method for surveillance and outbreak investigation.
Dr Keith Jolley is a bioinformatician working at the Department of Biology, University of Oxford, with Professor Martin Maiden. He has a PhD in Biochemistry from the University of Bath where he graduated in 1996. Following a PostDoc in Southampton, Keith moved to Oxford in 1998 to work on molecular typing of meningococcal carriage populations and gained programming and bioinformatics experience. Keith has been developing and overseeing the PubMLST website and the underlying molecular typing and genomic platforms for over 20 years (BIGSdb being the current iteration). He is also the co-developer of the ribosomal MLST scheme that can be used to identify and type all bacteria. The PubMLST website that he oversees hosts molecular typing schemes for more than 130 microbial species, serving as a nomenclature server for many third-party applications, and contains structured curated data for over 1.2 million bacterial genomes.
Professor Jason King is Advanced Vice-Chancellors Fellow and Royal Society University Research Fellow at the University of Sheffield.
Professor Robert A. Kingsley is a Professor of Microbiology at the University of East Anglia and a group leader and Principal Investigator at the Quadram Institute in Norwich, UK. His team’s work combines bioinformatics, genomics, molecular microbiology, and models of infection to investigate interactions between pathogens, the environment, and the host. Genomic diversity of bacterial pathogens and associated microbial and viral communities are studied to understand the function and evolutionary history of genes important to disease, transmission, drug resistance and phage sensitivity. Computational analysis generates hypotheses that are tested using classical microbiology, molecular genetics and models of infection. Our work has shed light on how new pathogens emerge and evolve, how epidemics spread and factors affecting risk to human and animal health. The long-term objective is to use this insight to improve policy decisions and in the rational design of intervention strategies, surveillance, and risk assessment of foodborne pathogens.
Dr Philippe Lehours has held a professorship at Bordeaux University Medical School and Bordeaux University Hospital since 2016. He is a pharmacist and holds a PhD in microbiology. During the period 2016-2021 he led an INSERM team focused on understanding the pathophysiological mechanisms of gastric carcinogenesis induced by Helicobacter pylori infection. Since 2017, he has headed the National Reference Center for Campylobacter and Helicobacter (NRCCH). At the NRCCH, he has routinely developed the sequencing of mainly Campylobacter genomes in order to study their phylogeny, resistome and virulome, and identify source attribution markers. NGS-type approaches directly from biological samples are also under development.
Professor Nick Loman works as Professor of Microbial Genomics and Bioinformatics in the Institute for Microbiology and Infection at the University of Birmingham. His research explores the use of cutting-edge genomics and metagenomics approaches to the diagnosis, treatment and surveillance of infectious disease. Nick has so far used high-throughput sequencing to investigate outbreaks of important Gram-negative multi-drug resistant pathogens, and recently helped establish real-time genomic surveillance of Ebola in Guinea. His current work focuses on the development of novel sequencing and bioinformatics methods to aid the interpretation of genome and metagenome scale data generated in clinical and public health microbiology.
Professor Martin Maiden is a molecular microbiologist. He attended the University of Reading (BSc, Microbiology with subsidiary Chemistry) as an undergraduate and the University of Cambridge as a Postgraduate (PhD, Biochemistry) and an MRC Training Fellow. He worked for nine years in the NHS (National Institute for Biological Standards and Control), including a one-year sabbatical in Germany (Max Planck Institut für Molekulare Genetic). He joined the University of Oxford in 1997 as a Wellcome Trust Senior Fellow. He has been a faculty member (Departments of Zoology and Biology), College Fellow (Hertford College), and Professor since 2004 and was Senior Proctor of the University of Oxford 2019-2020. From October 2024 he shall serve as Head of the Department of Biology, University of Oxford. For forty years he has applied molecular approaches to studying bacteria, with an emphasis on translation to infectious disease control, especially vaccination, with a strong emphasis on open access data. He is a committed educator and academic administrator.
Professor Alison Mather is a Group Leader at Quadram Institute Bioscience (QIB) and Professor of Microbial Genomic Epidemiology at the University of East Anglia (UEA); she is also the Head of the Microbes and Food Safety Institute Strategic Programme at QIB. Her research interests are to understand the origins, evolution and transmission of foodborne bacteria, with a focus on bacteria that are resistant to antimicrobial drugs. To do so she uses a variety of bioinformatics and statistical techniques, including short read, long read, and metagenome sequencing approaches. By defining and quantifying the importance of different sources of bacteria and AMR, the goals of her research are to address gaps in fundamental knowledge, identify points where interventions may be most effectively applied, and inform policy and surveillance.
Dr Jacqui McElhiney is Head of Food Protection Science and Surveillance and leads Food Standards Scotland’s science and evidence strategy and works closely with our Chief Scientific Advisor to provide assurance over our research and risk analysis processes. Jacqui worked for the Food Standards Agency in Scotland for 10 years as a senior scientific advisor in food safety. Her career followed a PhD in environmental science and 5 years of post-doctoral research in molecular microbiology and biochemistry. In her current role, she leads a team of scientific advisors who are responsible for all of FSS’s research, surveillance and risk assessment activities relating to food safety and food authenticity. She also provides oversight for data science and social research support across all areas of FSS’s remit.
Dr K. Marie McIntyre is the NUPAcT Fellow in Translational Food Safety in partnership with the FSA. Marie has over 20 years’ experience in research on infectious diseases impacting human and veterinary public health. She worked as an epidemiologist on scrapie and atypical scrapie at the Pirbright Laboratory, then moved to Health Protection Agency (now UK HSA) to work as Advanced Healthcare Scientist, followed by undertaking post-doctoral research on veterinary epidemiology and One Health approaches. In her Partnership Fellowship, she provides policy-appropriate ‘living’ evidence collation and synthesis, and risk assessment tools and mechanisms to aid the Food Standards Agency’s role in ensuring food for consumption is safe from foodborne disease and food supplies are sustainable. Marie is Co-Director of the AMAST (AMr in Agrifood Systems Transdisciplinary) Network, lead for Newcastle University of the National Institute for Health Research Health Protection Research Unit in Gastrointestinal Infections (2025-2032) and theme lead for the “Improving the evidence base for prevention of gastrointestinal infections” theme.
Alan McNally is a Professor in Microbial Genomics at the University of Birmingham and works on the evolutionary genomics of pathogenesis and antimicrobial resistance in bacterial pathogens. He has also been funded by the European Union, Medical Research Council and Royal Society. He has a strong belief in the collaborative nature of genomics research with active collaborations in the UK, China, Germany, France, Vietnam, and the US. During the COVID-19 outbreak, he was seconded to the Milton Keynes Lighthouse Lab as Infectious Disease lead at the Government’s first flagship COVID-19 testing facility. Launched on 9 April 2020, the Milton Keynes Lighthouse Lab was the first of three Government ‘mega-labs’ to be set-up across the UK, vastly increasing testing capacity and allowing tens of thousands more patient samples to be processed each day.
Dr Anais Païnset is a Lead for Bioinformatics at the UK Health Security Agency.
Professor Sascha Ott is an expert in developing targeted computational methods addressing biomedical needs. He is leading a research group at Warwick Medical School and a Bioinformatics service unit called Bioinformatics Research Technology Platform (RTP) with currently seven permanent staff members. His lab developed the “Wellington” footprinting methodology and software for high-throughput open chromatin data sets (Nucleic Acids Research 2015) which has found wide impact in cancer research, immunology, and a range of other biomedical settings, and a new method for the analysis of DNA methylation data which led to the hypothesis that stem cells may be depleted in the endometrium of recurrent miscarriage patients. His lab contributed to the design of the urine-based bladder cancer test GALEAS Bladder marketed by Informed Genomics. Since the retirement of Prof. Mark Achtman, Sascha has been responsible for the maintenance and development of the EnteroBase system by the Bioinformatics RTP team. He is PI on the BBSRC-funded PhytoBacExplorer project, developing an EnteroBase-like resource for the phytobacterial community. He also currently holds a joint Wellcome Trust Investigator award with Prof. Jan Brosens, and is co-investigator on one CMMI-EPSRC and one MRC-funded project.
Aleisha Reimer is Chief of Innovation and Application Development Section at the Public Health Agency of Canada’s National Microbiology Laboratory. Her section supports the reference, diagnostic, surveillance, and outbreak response functions of the Division of Enteric Diseases through the development and implementation of laboratory and bioinformatic tools and applied research in the areas of bacterial genomics, clinical and public health metagenomics, and artificial intelligence. She led the development and implementation of genomics for PulseNet Canada beginning in 2010. Current priorities include the development of Canada’s new Genomic Surveillance Platform and culture-independent subtyping approaches for enteric bacterial pathogens.
Associate Professor Torsten Seemann is a world-renowned Bioinformatician who has developed cutting edge analysis approaches to enhance the use of genomic data for Discovery Science and Public Health. He is the Lead Bioinformatician at the Centre for Pathogen Genomics and the Director of Bioinformatics at Doherty Applied Microbial Genomics. His expertise includes the management of hardware and the design of software analysis infrastructure required to interrogate pathogen genome data to understand pathogen evolution, transmission and drug-resistance. He also led national and international bioinformatic analysis for SARS-CoV-2 during the COVID-19 pandemic as well as the development and deployment of Australia’s first real-time data-sharing and national genomics surveillance platform, AusTrakka. Torsten has been a significant contributor to international genomics standards through the Public Health Agency for Genomics Epidemiology (PHA4GE) and has recently been appointed as Lead Bioinformatics for the US CDC’s PulseNet International Asia-Pacific Initiative.
Dr Eric L. Stevens is an International Policy Analyst in the Office of the Center Director at the Center for Food Safety and Applied Nutrition. He received his Ph.D. in Human Genetics and Molecular Biology from The Johns Hopkins School of Medicine with an emphasis on human population genetics and estimating genetic relatedness. Dr. Stevens works closely with international organisations and our foreign regulatory counterparts on food science and safety issues. Additionally, Dr. Stevens is the Codex Alimentarius manager for CFSAN and helps coordinate FDA’s participation with the US Codex Office and at Codex Committee Meetings.
Dr Adriana Vallejo-Trujillo is a researcher in WGS of microbial pathogens at the Roslin Institute, University of Edinburgh.
Dr Koji Yahara is a Group Leader at the Antimicrobial Resistance Research Center, National Institute of Infectious Diseases (a WHO Collaborating Center for AMR surveillance and research) in Japan. He is a bio- and medical-informatician responsible for Japan’s national AMR surveillance systems. He earned his Ph.D. in Biostatistics through a collaborative program bridging biostatistics and infectious disease epidemiology. In 2012, he began a long-term research stay in Germany (at the Max Planck Institute) and the UK (collaborating with Swansea, Oxford, and Imperial College London) as a government-funded JSPS Research Fellow from the University of Tokyo, contributing to international research projects on bacterial population genomics. Upon joining the National Institute of Infectious Diseases in April 2016, he assumed responsibility for the information technology of the Japan Nosocomial Infections Surveillance (JANIS), a comprehensive national AMR surveillance program. Since 2019, his team has been developing genomic surveillance programs and studies, as reported in Genome Medicine (2021) and Nature Communications (2023). His primary focus lies in computational and statistical analyses of genomic and epidemiological data related to infectious diseases, as well as in the development and management of information technology programs and databases for the national surveillances. He also serves as an Editor for Microbial Genomics.