Introduction
As part of the Innovation Research Programme (IRP), the Food Standards Agency (FSA) and Food Standards Scotland (FSS) carried out several expert elicitation events to explore the most common challenges applicants face when preparing precision and biomass fermentation (PF) applications for assessment. The focus topics of these events were selected through a previous internal scoping exercise that identified recurrently challenging areas for applicants as: bioinformatics, protein digestibility related to nutritional quality, allergenicity and clarity on the identity and composition of the product.
From the output of these expert elicitation events, the FSA and FSS have compiled a list of guidance for readying PF applications that is presented in this report.
Scope
This section aims to inform on the conceptual framework used for the expert elicitation events and the products in scope of this supplementary guidance.
The Food and Agriculture Organisation (FAO) has not adopted a single, official definition for precision fermentation, however, in general terms, precision fermentation can be described as the use of engineered or selected microbes, grown in controlled conditions, to make specific purified molecules (proteins, lipids, vitamins, enzymes, etc.) traditionally sourced from plants or animals.
At the time the expert elicitation events were carried out, the FSA used the following two definitions to frame the discussion:
-
Working definition of PF: Precision fermentation is considered to be a technology that creates essential food components, like proteins and enzymes, through use of genetically modified microorganisms to produce specific functional components.
-
Wider definition of PF: where whole microorganisms are reproduced in a bioreactor / closed system and refined for their fat or protein content (includes the working definition, as well as biomass fermentation).
The definition of PF as interpreted by the FSA has been updated ahead of any future work. The most up to date Policy position on PF can be found here: Precision Fermentation Business Support Service (BSS) | Food Standards Agency
Products in scope of this guidance are those that meet the definition of a novel food as per Article 3 of EU Regulation 2015/2283 produced by precision or biomass fermentation. To support this regulation, the Food Standards Agency (FSA) follows the 2016 EFSA guidance on novel foods.
Organisms in scope of this guidance were selected non-genetically modified (GM) microorganisms and category 1 and 2 GM microorganisms (where no GM DNA content is present in the final product), covering bacteria, yeasts and filamentous fungi. Out of scope microorganisms include microalgae, plants, animal cells, category 3 and 4 GM and precision bred organisms (PBOs). This list is not exhaustive, and some guidance points listed in this document may be relevant to excluded microorganism types.
This supplementary guidance document does not replace the FSA/FSS guidance or the EFSA novel foods guidance for preparing novel food dossiers, and applicants should continue to follow those guidance documents closely. While this document outlines key considerations for novel foods produced through PF and biomass fermentation, it is not an exhaustive list. The guidance points listed here focus primarily on the novel food assessment areas that were highlighted as recurring issues and therefore explored through the expert elicitation events (identity, bioinformatics, composition, protein digestibility and allergenicity).
Depending on the specific characteristics of the novel food under assessment, the FSA/FSS may also request information in addition to what is outlined in this report. Applicants should prepare their dossier, drawing a clear narrative, and interpreting their results considering the specific characteristics and conditions of use of their product. Following this guidance does not necessarily guarantee that a product will be authorised. It is a matter for Ministers to determine whether or not to authorise a novel food produced by precision fermentation and there are a variety of case-specific factors which may inform that decision.
Supplementary guidance
1. Identity – Bioinformatics
General considerations
-
EFSA’s 2024 guidance on whole genome sequence analysis of microorganisms intentionally used in the food chain is helpful background information for applicants to use for detailing genome sequence requirements.
-
Applicants should be aware that whilst bioinformatics analyses are helpful to support safety assessments, these are a complimentary part of the wider evidence package and should be used by the applicant to provide context for the full application.
-
The examples provided are not exhaustive, and it is essential that applicants determine the specific requirements applicable to the characteristics and conditions of use of their product.
-
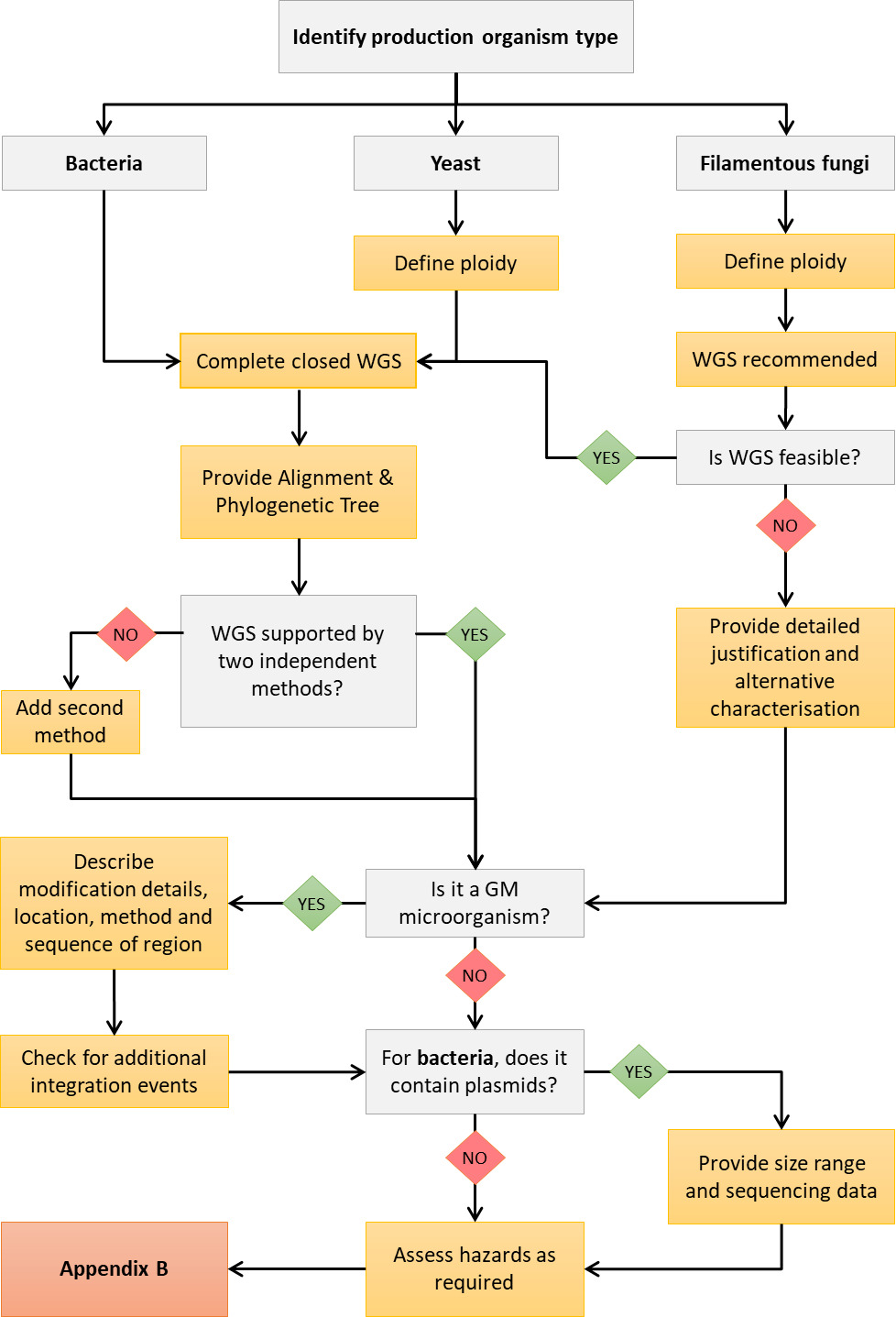
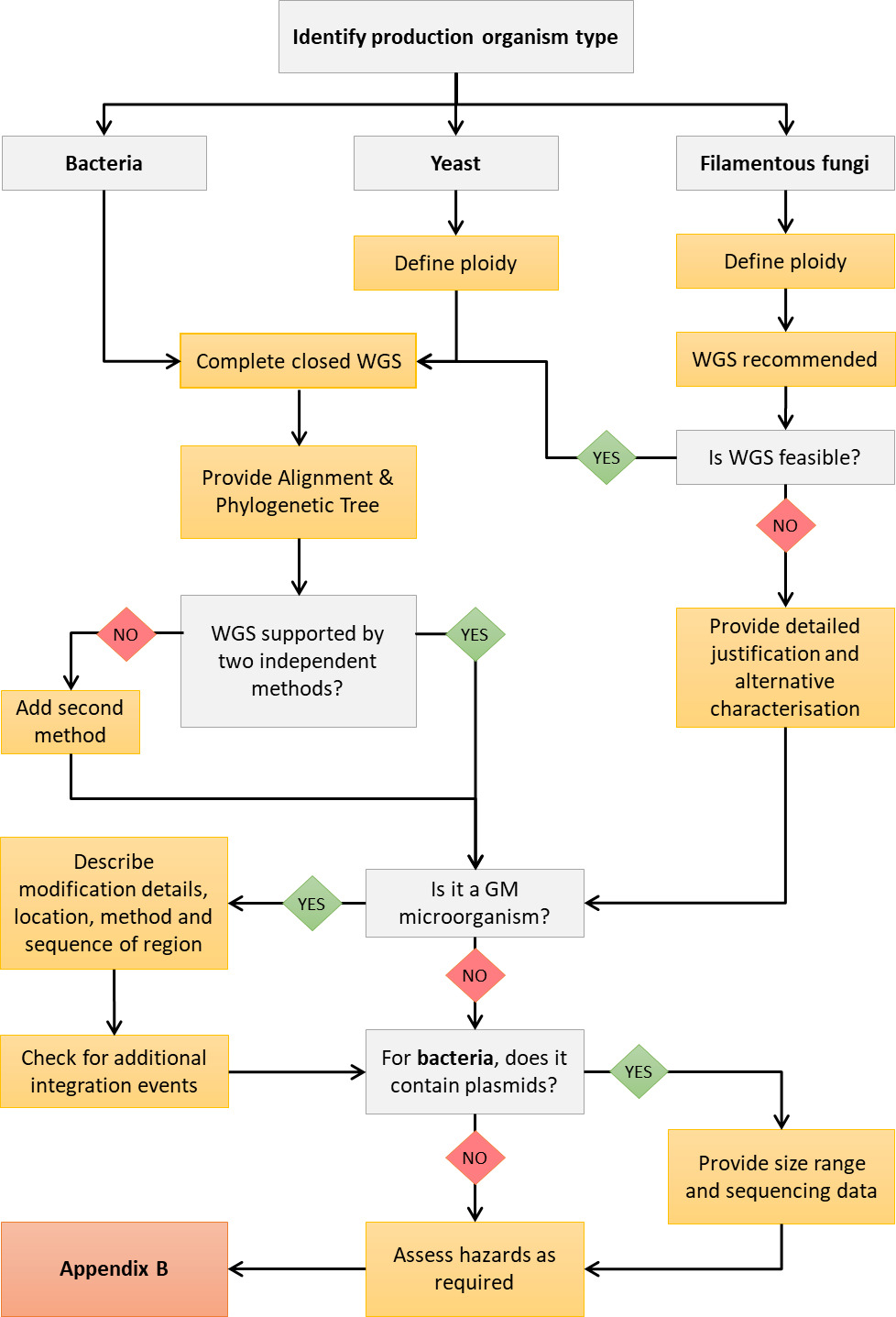
Appendix A shows a decision tree to help applicants determine where to start when characterising their production organism through whole genome sequencing (WGS).
Whole genome sequencing guidance
-
As a general point, applicants are advised that the less well-known an organism is within the context of novel foods, the more helpful a WGS may be to establish their safe use.
-
If a production organism has QPS (qualified presumption of safety) status, applicants should provide evidence that this is the case, and whether the QPS status was granted for the organism as a whole or only as a production organism. Please note that the genome-based evidence described in this report should still be provided regardless of the QPS status of the organism.
-
For bacteria and yeasts, a complete closed WGS is considered as standard, and justification should be provided where this is not supplied. It is expected that, for bacteria, plasmid and non-nuclear DNA is accounted for as part of the sequencing exercise.
-
Plasmids often encode compounds contributing to key safety risks, including antimicrobial resistance (AMR), toxins, and virulence factors. Applicants should state whether the production strain contains plasmids and, if so, provide size range estimates and sequencing data for these, using adapted sequencing methods that adjust for sensitivity for small plasmids, and dual approaches (long and short read coordinated sequencing) for large plasmids.
-
For fungi, the ploidy level should be defined. Sequencing of non-nuclear DNA is not normally required.
-
For filamentous fungi, while whole genome sequencing is still recommended to characterise the organism, it is recognised that this may not always be feasible. Where this is the case, applicants should provide justification.
-
Where a WGS is expected, identity should be supported using whole-genome approaches, ideally with at least two independent methods. For fungi, multi-gene phylogenetics (e.g., BUSCO gene sets) and/or whole-genome approaches (k-mer/SNP-based) are appropriate. Where only a partial sequence is generated, applicants should specify the completeness of the sequence and describe how missing regions were identified or reconstructed using related reference genomes
-
To identify and characterise a production organism, a molecular phylogenetic tree should be provided. To assist the FSA in judging the quality of a phylogenetic tree, applicants should provide alignments together with an accompanying narrative describing how it was constructed, including information about which tree-building method was used (e.g. what are the sister groups, why particular reference strains have been chosen, why the tree is rooted in a certain way, and tree-building approach). A Maximum Likelihood Estimation (MLE) method is recommended.
-
For genetically modified organisms (GMOs), details of where genetic modification has taken place, how the modification was achieved, the sequence of the modified region and precise information on whether genetic information is integrated into the genome, or whether it is present on a plasmid or plasmids, should be provided.
-
Applicants should submit evidence on whether additional integration events (in addition to those intended) have occurred in the GMO, and if so, a complete analysis should be provided with evidence on the likely impact of these, for example on downstream gene expression, or on disruption of gene function.
Suggested databases and formats
-
Applicants should use well-established databases and tools for genome analysis, and these should be relevant to the type of microorganism and the nature of the hazard under evaluation. Applicants are encouraged to use the best available tools.
-
In Tables 1 and 2 (Appendix B), there are examples of tools and databases which exemplify best practice when analysing a genome to identify risks. It is the applicant’s responsibility to identify the appropriate databases and analysis tools to characterise the specific qualities of their production organism. These resources support risk assessments such as allergenicity, toxicity, and AMR risk.
-
Appendix C contains further information on recommended sequencing data file formats.
-
Applicants can refer to the EFSA statement on the requirements for whole genome sequence analysis of microorganisms intentionally used in the food chain, for suggestions of minimum parameters to use and suitable thresholds.
2. Production process
-
Regarding microbiological safety, detailed process information, including purification steps and kill-step validation data, is required to provide additional evidence of the removal of host proteins and viable genetically modified microorganisms (GMM). Validation should include information on the sampling plan, heat treatment conditions (temperature and retention time) linked to microbial reduction and HACCP documentation.
-
Providing evidence of kill-step validation and DNA degradation is particularly important as part of the production process for biomass products. Applicants should provide batch-level evidence, typically from 5 batches.
-
To account for the potential genetic drift of the production strain, and as part of the production process, applicants should show evidence of good laboratory practice when banking and handling the strain. Information should be provided on how many fermentation cycles the same inocula is used for, before restarting with a new inocula. Whole genome sequencing would not be expected to be used to evaluate genetic drift of the production strain.
3. Compositional data
Impurities
-
Applicants should test for the absence of the production organism in the final product. After initial quantification, shotgun proteomics are recommended to identify any residual proteins derived from the production organism.
-
Where WGS data is available, proteomic analysis for peptide identification is recommended. This process should be used to confirm the absence of unintended peptides and proteins.
-
When evaluating residual DNA presence in the final product, proportionate requirements based on product type and exposure should be considered. For example, although the presence of residual DNA risk is reduced for highly purified products, verification is still required. For biomass-based products, the risk of residual DNA being present is potentially higher and as such, applicants should apply more stringent checks.
Stability
- Regarding shelf-life stability, for food substances produced through PF or biomass fermentation that possess specific functional properties, it should be verified that these properties persist during storage.
4. Proposed uses and use levels and anticipated intake
-
Nutritional and intended use assessment for PF products should start with existing guidance, but be adapted for high-concentration uses, population specific-risks, and compositional variability. This should be applied on a case-by-case basis, and applicants should take into consideration the multiple potential uses of the food product.
-
Applicants should take into consideration the intended use and the target population for PF products when assessing and interpreting results for their dossier, particularly for vulnerable groups.
5. Nutritional information – Protein digestibility assays
Proportionate Approach
-
The decision on whether digestion assays are required should be determined using a proportional, tiered approach that aligns the evidence with the ingredient’s intended use, proposed use levels, the food categories in which it will be used, and expected exposure, with explicit consideration of vulnerable populations (e.g., infants, young children in relevant categories, older adults, and individuals with special nutritional needs). Information will be most useful where the product provides a source of protein in the diet in order to assess nutritional disadvantage.
-
Low-inclusion techno-functional uses are considered around ≤5% inclusion of the final product, whereas uses as source of protein are considered around ≥12% inclusion. However, this should be treated as an illustrative benchmark with decisions driven primarily by the ingredient’s actual intended role (e.g., techno-functional, nutritional) and the resulting exposure.
-
Proteins used solely for techno-functional purposes at very low inclusion levels (e.g. nutritional enzymes, emulsifying peptides, or ice-structuring proteins) generally do not need a nutritional digestibility assay. At these low use levels, the contribution to overall dietary protein intake is not significant enough to contribute meaningfully to diet requiring nutrition-focused digestion testing. This does not, however, preclude the need for digestion data to peptide level to support allergenicity risk assessment.
-
Where an ingredient is intended to act as a major source of protein or to replace established protein sources (e.g., meat, milk, or egg analogues, protein supplements, or high-protein microbial biomass), a digestion assay should be provided. In such circumstances, the assay should provide the clearest evidence of the contribution this would play to the diet in order to assess nutritional disadvantage, with particular attention to vulnerable populations.
-
Nutritional equivalence for specific proteins should be demonstrated at the level of key biochemical functions, not just sequence or total protein.
Preferred nutritional endpoint
-
DIAAS (Digestible Indispensable Amino Acid Score) should be the primary score for assessing protein quality. Other metrics such as Protein Digestibility-Corrected Amino Acid Score (PDCAAS), Digestible Indispensable Amino Acid Ratio, amino acid profiles and nitrogen data can also provide useful supportive data, but DIAAS is preferred.
-
The nutritional digestibility assessment should start with the ingredient itself, using a validated digestion method, preferably in vitro digestion method such as INFOGEST to generate the nutrition endpoint (DIAAS).
-
In vitro testing is highly recommended over in vivo methods. Pig models are recognised as closer to human GI physiology, however, pig studies should be reserved for specific, unresolved questions that in vitro methods cannot answer or to validate in vitro methodologies. The use of small rodent studies is discouraged.
-
A study in a representative matrix may be required where assessment of the ingredient alone is not representative of the intended use:
-
The ingredient is unprocessed/raw and likely to contain antinutritional factors or toxins that processing would modify, or processing is expected to materially change digestibility.
-
The main intended category imposes a matrix or consumer preparation that plausibly alters protein accessibility (e.g., under cooked protein not reaching adequate internal temperatures).
-
There is a credible exposure in vulnerable groups (toddlers, elderly, or special nutritional needs) where small shifts in digestibility could have nutritional impact.
-
-
In such cases, a single representative processed matrix should be selected and scientifically justified (time/temperature, typical consumer preparation, intended use).
Performance criteria needed
-
Applicants should run at least 3 technical replicates, include reference proteins, verify enzyme activities before the method is applied. The test material used in the digestibility assay would ideally be one included in the compositional analysis in the dossier, so its full composition is known.
-
Each experimental run should include at least one “high digestibility” reference (e.g., milk/casein or skim milk powder) and ideally one “moderate digestibility” reference (e.g., soy). References should be digested alongside the test ingredient (same fluids, enzyme activities, times, temperatures, sampling) and analysed with the same LC-MS/MS (or an equivalently validated technique) amino acid workflow used for the test item. It should be noted that literature values cannot substitute for in run references because the assay conditions and analytics drive the outcome and therefore need to be identical to conditions of the test ingredient.
-
In addition to appropriate references in every run, appropriate comparators should be included– the proteins that are intended to be replaced by the newly-generated ingredient (e.g., casein for dairy analogues) and nutritional equivalence in intended uses should be clarified.
-
The INFOGEST static protocol is recommended as the default in vitro method for protein digestibility assessment. Further information on the standardisation of assay methods can be found in Appendix D.
6. Allergenicity
-
For purified proteins produced from a GMM, it is recommended that the applicant provides a full amino acid list to confirm the identity of the intended protein. However, if differences arise in thermal denaturation or functional tests between the recombinant protein and its natural equivalent, characterisation of the proteins’ folding and tertiary structure is recommended to determine functionality and allergenicity.
-
It is recommended that an evaluation of folding and post-translational modifications (PTMs) is undertaken. If folding or PTMs differ significantly, then more advanced allergenicity tests (e.g., IgE-binding studies) may be warranted. Conversely, if equivalence is demonstrated, extensive allergenicity testing may not be proportionate.
-
Requirements will depend on whether the protein is newly-generated or a known allergen, and on the intended functional role. For proteins that have nutritional or functional roles, the applicant should demonstrate that the recombinant protein retains this functionality. This may require additional tests beyond composition, such as mineral-binding assays.
-
Applicants should identify the protein being replaced and provide evidence demonstrating that its micronutrient-binding function is retained. Where nutritional fortification is applied, applicants should also show that micronutrient bio-accessibility and absorption remain adequate under realistic consumption conditions.
-
It is recommended that applicants begin with structural and functional characterisation. Intact mass analysis is suggested as an important first step to confirm the protein identity and detect unexpected modifications.
-
The allergenicity of the newly expressed protein should be considered in the form in which it will be consumed and any host proteins that may accompany it. For each of these aspects, applicants should evaluate the risks posed by the protein to the existing allergic population:
-
If the target protein is a known allergen, the structural characterisation should be done on a food product relevant to the conditions of use, together with analytical data showing consistency across batches.
-
If the newly produced protein is an allergen, and it is shown to be structurally equivalent to the existing allergen, the allergenicity risk assessment should be carried out as if it was an existing allergen.
-
If the newly produced protein is not structurally equivalent to the existing allergen, the risk assessment should follow the steps for new proteins, which include: history of use and allergenicity; bioinformatic analysis; in vitro digestibility testing; targeted serum IgE testing and other next steps; and assessment of allergen exposure. A tiered approach is recommend to ensure data generated is relevant to the product seeking authorisation.
-
-
Where bioinformatic sequence analysis is undertaken to help inform the allergenicity assessment, it is recommended that interpretation of the results exceeds raw sequence similarity and considers factors such as clinical relevance of the allergen source and whether the allergen is expressed in the vegetative state or only in spore form.
-
Applicants are reminded that digestibility assays for allergenicity risk assessment are distinct from those for nutritional quality. In vitro digestion models such as gastric and early duodenal phases are relevant for allergenicity risk assessment.
-
For clinical (serum) studies, extensive allergenicity testing (e.g., IgE-binding studies) may not be necessary if the protein is structurally and functionally equivalent to its natural analogue.
Acronyms
Appendices
Appendix A: Decision tree for WGS analysis
Appendix B: Example databases for WGS
Appendix C: Recommended sequence data file formats
Applicants should provide sequencing and alignment data in standard, widely accepted formats to ensure interoperability and compliance. Recommended sequencing data file formats, based on the sequencing techniques, include:
Sanger sequencing data
-
Raw reads:
-
AB1 format (.ab1 or .abi). mandatory if Sanger sequencing was used.
-
Each file represents a single read; files should be clearly named and organised to indicate their origin and purpose.
-
-
Assembly and alignment:
-
FASTA or FASTQ, to finally assemble the sequence
-
These files should be in plain-text format (parsable), not embedded solely in PDFs.
-
Next-Generation Sequencing (NGS)
-
Raw reads:
-
FASTQ – mandatory for NGS platforms (e.g., Illumina, PacBio)
-
Typically provided in compressed form (.fastq.gz), but the uncompressed format is plain text (parsable).
-
-
Alignment files:
-
BAM format (Binary Alignment Map) – strongly preferred for efficiency and compatibility.
-
SAM format (Sequence Alignment Map) – acceptable but not preferred due to large file size.
-
If alignment represents reads mapped to a reference, the reference sequence file should also be provided.
-
-
Paired-end reads:
-
Forward (R1) and reverse (R2) files should contain the same number of reads in matching order.
-
Interleaved FASTQ format is acceptable if alternating R1/R2 reads are clearly structured.
-
Annotated Gene and Protein Sequences
-
Annotated genome
- General feature format (.gff file).
-
Gene sequences:
-
GenBank or EMBL format – mandatory for annotated DNA sequences.
-
Should include all features (e.g., CDS, promoters) in formal specification.
-
-
Protein sequences:
- FASTA format – mandatory for all proteins encoded by the event sequence.
Post-sequencing data Integrity and Verification
-
Checksum files:
- MD5 checksums are strongly recommended for all major folders to verify data integrity.
File structure
-
Include a README file explaining folder contents and any deviations.
-
Information should be structured in a way that is accessible and meaningful to support timely review by the FSA.
Interpretation and Documentation
-
Applicants should interpret and explain all analyses in the dossier, including:
-
Database versions and software tools used.
-
Methodology for sequencing, alignment, and risk assessments.
-
Any deviations from standard practices.
-
Extension of use
- Depending on the nature of the extension of use application, a new genome sequence may be required.
Timeframe
- All bioinformatics analyses should be conducted within one year prior to the date of application submission.
Appendix D: Standardisation of assay methods
-
For assessing nutritional protein quality and determining DIAAS, the physiologically relevant analyte is the soluble small intestinal digest fraction, comprising soluble peptides and free amino acids that are available for absorption in the small intestine. Measuring this fraction ensures the endpoint reflects material that can be absorbed and used for amino-acid scoring.
-
Given the inherent heterogeneity of digesta, reproducibility should be improved by using independent reaction vessels for each time point rather than repeatedly subsampling from a single vessel. Where required by the analytical endpoint, the digest should be fractionated into soluble and insoluble phases and the fractionation methods described. The insoluble fraction supports mass-balance checks and method verification, whereas the soluble fraction representing absorbable material is the one analysed for DIAAS.
-
The recommended analytical endpoint for nutritional analysis is indispensable amino acid profiling by LC–MS (liquid chromatography-mass spectrometry) (or an equivalently validated technique) performed on the soluble fraction to enable robust DIAAS calculation. OPA and similar colourimetric peptide assays may be useful for screening or trending; however, they are insufficient to compute DIAAS and are susceptible to methodological variation (e.g., differing responses to peptide length/composition).
-
Standardised validated amino acid analysis should account for residues and processing induced modifications (e.g., Maillard modified lysine), apply appropriate hydrolysis conditions, and incorporate mass balance assessments to verify recovery and completeness. Use of LC–MS improves specificity and selectivity compared with nonspecific colourimetric approaches.
-
A clear validation package should be provided for amino acid analysis (e.g., precision, accuracy, recovery, linearity, limits of detection/quantification) and describe sample handling (collection, fractionation, stabilisation, and storage) to demonstrate control of pre-analytical variability.
-
Routine verification of enzyme activities before each experiment is crucial, due to potential loss of activity during storage and handling. Enzyme dosing should be reported in terms of measured activity units rather than nominal amounts from bottle labels, and the verification procedure (assay conditions, substrates, units, and frequency) should be documented. All critical reagents (e.g., bile salts/surfactants and buffers) should be qualified for identity, potency, and suitability for purpose, with lot numbers recorded and any acceptance criteria and stability controls described to demonstrate consistency across runs.
-
Each digestion run should include well characterised reference proteins processed in parallel under identical conditions to reveal method bias and support comparability of outputs (see 6.4.2).
-
For protein ingredients rather than complex foods, proportionate adaptations to the INFOGEST protocol are acceptable where justified by composition and intended use:
-
In the oral phase, salivary amylase may be omitted when starch is negligible, while the simulated salivary fluid is retained to provide dilution and ionic context. Where starch content is appreciable, amylase should be included; as a pragmatic guide, a threshold band of 10% starch in the test material was discussed, subject to sense checking against real applications and product specifics.
-
In the gastric phase, gastric lipase may be omitted for low lipid protein ingredients. Where lipids are substantial, lipase should be included, recognising that fatty acids and protein–lipid interactions can influence proteolysis and micellarisation. Most protein ingredients are relatively low in lipids, but the choice should be composition led rather than assumed.
-
In the intestinal phase, pancreatin is appropriate for the nutrition workflow, supporting endpoint amino acid analysis of the soluble fraction. For allergenicity and certain toxin/anti nutritional factor investigations, where the detection and tracking of intact proteins or specific peptide fragments are critical, purified proteases may be preferred to reduce background and to facilitate readouts such as SDS-PAGE densitometry and antibody-based methods.
-
-
Any deviation from the standard INFOGEST protocol or other validated method (e.g., omission of an enzyme, alteration of phase conditions) should be explicitly justified using composition data (starch, lipid, fibre) and relevant literature and documented alongside the digestion run (including reagent lots, activities, and acceptance criteria) to maintain transparency and reproducibility across experiments.
-
Digestion experiments should include at least three independent replicates per condition. Test material should be taken from a batch included in the product’s compositional analysis so that the digestion data can be interpreted in the context of the material placed on the market. If replicate digestions are conducted across different batches, the associated reference controls should accompany each batch so that any batch related bias is visible and can be addressed.
-
Transparent DIAAS calculations should be provided, making clear how the values were derived from the amino acid data. The robustness and comparability of the digestion workflow should be demonstrated through independent replication (at least three digestions per condition), verification of enzyme activities before dosing, and the inclusion of well characterised reference proteins processed in parallel to reveal any systematic bias and to support interpretation of results.
-
Where methods other than the INFOGEST static protocol (including dynamic systems) are used, applicants are expected to:
-
Justify fitness for purpose, explaining why the chosen system is appropriate for the intended endpoint(s).
-
Provide transparent calculation methods showing how model outputs are converted to nutrition endpoints, explicitly describing sampling schemes (e.g., time integrated or cumulative) and their potential effects on values.
-
Run appropriate reference proteins in parallel to detect systematic bias and enable method to method comparability.
-
Present evidence of validation, such as inter laboratory comparison and/or correlation to suitable in vivo datasets.
-
Reconcile any discrepancies between the alternative method and the INFOGEST static protocol, explaining observed differences and their implications for assessment.
-
-
For DIAAS and exposure assessments, the amino acid composition should be used to derive protein content for calculations, rather than relying solely on total nitrogen with default conversion factors (e.g., Kjeldahl/Dumas). This is particularly important for newly-generated proteins (such as microbial/precision fermentation materials) where non-protein nitrogen can bias totals if default factors are applied. While labelling considerations may require the use of standard nitrogen to protein factors, applicants should present the amino acid derived protein value for scientific assessment and be prepared to explain any divergence between the two approaches.
-
Where accreditation is not available, applicants should provide an auditable method-validation package and be prepared to permit inspection/audit if required. The package should demonstrate that the workflow is robust, reproducible, and relevant to the regulatory requirements. Applicants should submit full protocols, enzyme activity records, instrument methods, raw data files, calculation workbooks, and statistical summaries (means, SD, CIs, outlier treatment).
-
Dossiers should report end-to-end processing conditions for each ingredient or material tested, including time, temperature, pressure, pH adjustments, moisture, shear, residence times, and any pre-treatments applied for microbiological control. “Unprocessed” ingredients that undergo concentration and/or drying should have these conditions reported, as such steps frequently involve heat exposure with meaningful effects on digestibility.
-
Distinctions should be drawn between products taken in discrete doses to increase intake (e.g., scoopable powders) and uses in which only small amounts of protein are incorporated into conventional foods for technological purposes, (such as structuring, emulsification, or enzyme-like activity) without an intention to supply protein nutrition.