















1. Acknowledgements
This work is funded by the PATH-SAFE program from the HM Treasury Shared Outcome Funds. The authors thank the microbiology staff of the clinical microbiology laboratories at the John Radcliffe Hospital in Oxford and the Singleton Hospital in Swansea for providing Campylobacter isolates from human disease. We also thank industry members and farmers for providing samples from livestock and poultry, and pet owners for providing samples from backyard chickens.
2. Lay Summary
Campylobacter is the most common cause of bacterial food poisoning in the UK. Although infection in humans is rarely fatal and usually resolves without treatment after 5-7 days, antibiotics may be needed for severe cases, people with a poorly functioning immune system, the young or the elderly. Disease caused by Campylobacter is a major burden to public health and causes economic loss in excess of £700 million (Daniel et al., 2020) each year in the UK. Furthermore, the problem is worsening as existing treatments for severe infections are becoming less effective with the rise of antimicrobial resistance (AMR). Indeed, Campylobacter were designated ‘high priority’ pathogens on the 2017 WHO watch list[1] for antimicrobial resistance (Veltcheva et al., 2025).
In this study, we determined the current levels of AMR amongst Campylobacter isolated from clinical patients (‘human disease isolates’) in Oxfordshire, which, as part of an ongoing 22-year study, have been previously shown to mirror national trends. We also included a small number of human disease isolates from Wales. In order to improve the accuracy of methods to predict the source of human infection, we tested Campylobacter isolates cultured from potential sources that are under-represented in national culture collections (for example, wild birds, cattle, sheep, horses, deer, free-range chicken and chicken breeder flocks). We DNA sequenced each of the Campylobacter isolates to identify genes that predict antibiotic resistance. In addition, DNA sequence data was used to improve modelling approaches to predict the sources of human infection and to assess potential risk of spread (‘spillover’) of antimicrobial resistant Campylobacter from agriculture to the environment. This study period covers the COVID-19 lockdowns (2020-2021), representing a natural experiment assessing the impact of restrictions affecting eating out, social gatherings and foreign travel. We continued to develop automated methods to identify antibiotic resistance genes, and to enhance typing methods to improve outbreak detection for human infection. Data were stored and analysed using PubMLST, a publicly available database which provides definitive naming systems for Campylobacter genes and variants for the global research community.
Key findings from the project are that resistance to two classes of antibiotics called ‘fluoroquinolones’ and ‘tetracycline’ with different modes of action has increased amongst human disease isolates since our last study in 2015-2018. Within the Campylobacter jejuni species, there are three closely related genetic lineages that have resistance approaching 100% to these antibiotics, and further research is needed to understand their high prevalence despite significant reduction of antimicrobial use on farms. The COVID-19 lockdowns with restricted eating out and travel did not impact the numbers or genotype distribution of Oxfordshire human infection. More than 80% of human disease is predicted to come from chicken, though it was not possible to determine precise routes of infection (e.g., undercooked meat versus cross contamination in the kitchen, home prepared versus commercially prepared food and takeouts, imported versus UK meat), and further research is recommended.
3. Executive Summary
Campylobacter (principally the species Campylobacter coli and Campylobacter jejuni) is the most common cause of bacterial gastroenteritis in the UK. Although human campylobacteriosis in healthy individuals is rarely fatal and usually self-limiting, the disease is a major burden to public health and causes economic loss in excess of £700 million. Campylobacter infection may also lead to sequellae such as Guillain Barré Syndrome (GBS), reactive arthritis (ReA) and irritable bowel syndrome (IBS). Furthermore, the problem is worsening as existing treatments for severe infections are becoming less effective with the rise of antimicrobial resistance (AMR). Indeed, AMR Campylobacter were designated ‘high priority’ pathogens on the 2017 WHO Bacterial Pathogen Priority List[2] for antimicrobial resistance.
The aims of this study were to i) determine the current levels of AMR amongst UK human disease and AgriFood isolates and ii) to characterise Campylobacter transmission routes in Agri-Food systems. Specifically, the aims were designed to update information and address missing knowledge gaps by including isolates from sources that have been previously under sampled, such as cattle, sheep, wild birds and free-range poultry. Intended outcomes of the project were to assess the risk to human health and effectiveness of ongoing interventions by comparing current levels of Campylobacter AMR with existing isolate collections.
Given the extremely high levels of human disease, reducing infection from any source will potentially lead to a significant reduction in the actual number of people infected. Genomic mechanisms underlying AMR were investigated using the fully curated data available on PubMLST. Resources were enhanced and integrated with the PATH-SAFE programme for efficient storage and sharing of data, and to enable further analysis of the impact of globally integrated food distribution systems.
In this study, we determined current levels of AMR (predicted from genotype) amongst Oxfordshire human disease isolates (2019-2024) that are representative of national trends, together with a small number of isolates from Wales (2012-2013). In addition, we whole-genome sequenced isolates from sources that are under-represented in national culture collections, such as cattle, sheep, deer, horses, wild birds, free-range broiler (meat) chickens and broiler breeder flocks. This will help to improve the accuracy and robustness of source attribution models, investigate the evolution of AMR genetic determinants, and assess ‘One Health’[3] interaction between people, animals and the environment. This study period covers the COVID-19 lockdowns (2020-2021), where, using genotyping data to compare before, during and after the pandemic, we were able to assess whether restrictions on eating out, social gatherings and foreign travel affected the source or transmission of Campylobacter variants causing human disease. We also continued to develop molecular typing methods, data curation and reporting of AMR genetic determinants using PubMLST, a publicly available database which provides definitive nomenclature for Campylobacter and other microorganisms, and enables storage and downstream analysis of isolate genomics and metadata.
A key finding from the study is that more than 80% of human disease caused by Campylobacter continues to be derived from chicken, demonstrated by predictive source attribution analysis. However, each of the under-represented host sources tested in this study, including pet/backyard chickens, wild birds and deer had potential to cause human disease. The generalist ST-21 and ST-45 clonal complexes and also the ruminant associated ST-61 clonal complex were common to a number of sources (cattle, sheep, deer, horse, starling, geese and the farm environment) and human disease, however further work is needed to elucidate transmission routes amongst these generalist clonal complexes. This study period covers the COVID-19 lockdowns (2020-2021), where we observed no change in the number or composition of Campylobacter variants in Oxfordshire human disease, despite restrictions with respect to eating out, social gatherings and foreign travel. It was not possible to determine precise routes of infection (e.g., undercooked chicken meat versus cross contamination in the kitchen, home prepared versus commercially prepared food and takeouts, imported versus UK meat) however, and further research is recommended.
Fluoroquinolone and tetracycline resistance has continued to increase amongst Oxfordshire human disease C. jejuni isolates since our last study in 2015-2018. In 2015-2018, 35.8% of Oxfordshire human disease isolates were fluoroquinolone resistant, (increased to 55.2% resistance in 2024), and 32.6% of isolates were tetracycline resistant in 2015-2018, (increased to 48.2% in 2024). Resistance amongst Oxfordshire C. coli isolates has for the most part fluctuated between 30-45% for fluoroquinolones and 20-40% for tetracycline over the past decade.
Co-resistance to both fluoroquinolone and tetracycline has been increasing since we began surveying Oxfordshire human Campylobacter disease in 2003, most commonly amongst C. jejuni, where 42.5% of isolates were predicted to be resistant to both antimicrobials in 2024. Particularly concerning are the ST-353, ST-354 and ST-464 clonal complexes commonly associated with chicken, that have resistance approaching 100% to fluoroquinolones and tetracycline. These clonal complexes were found only amongst poultry sources in the study but covered a diverse range from pet chickens to broiler breeder flocks, and from multiple farms where antibiotic selection pressure would likely vary. Further research is needed to understand their high resistance despite significantly improved antimicrobial stewardship on farms. The situation appears complex since fluoroquinolone and tetracycline resistance has been consistently linked with these particular Campylobacter lineages, implying there may be little fitness cost to the bacterium, and/or there is co-selectional advantage in the present conditions, whilst others such as ST-257 clonal complex have remained highly sensitive.
Levels of fluoroquinolone and tetracycline resistance were very low (<5% of isolates) amongst ruminants (cattle and sheep), and wild birds (geese and starlings), with the exception of tetracycline resistance amongst starlings. Note, the resistant isolates from geese and starlings belonged to clonal complexes and STs most commonly associated with livestock and not from either of the wild bird sources. The seemingly higher prevalence of tetracycline resistance amongst ‘starling’ isolates here is almost certainly an over-estimation due to the isolate selection bias that was necessary to cover the diversity of Campylobacter lineages. Rarer lineages consequently gave a higher signal than would occur when sampling a normal population. Overall, the results indicate that ruminants and wild birds pose a low risk of causing AMR Campylobacter disease in humans. The occasional livestock-associated resistant Campylobacter isolated from wild birds highlights the need for ongoing biosecurity and vigilance in maintaining low AMR in ruminant-associated Campylobacter lineages and preventing agricultural spillover to the environment (eg run-off from faecal waste into water courses).
Prevalence of macrolide (0.04% C. jejuni, 4% C. coli) and aminoglycoside (1.9% C. jejuni, 12% C. coli) resistance was relatively low amongst human disease Campylobacter isolates compared to fluoroquinolone and tetracycline resistance. It should be noted that erythromycin (macrolide) resistance was at the highest minimum inhibitory concentration (MIC) level for the isolates tested phenotypically, despite absence of the ermB gene. Presence of the recently discovered resistance enhancing efflux pump genetic determinants was variable amongst the isolates. Amongst the AgriFood and environmental isolates tested in this study (cattle, sheep, deer, extensively reared broiler (meat) chickens, broiler breeder flocks, horses, geese and starlings) no macrolide resistance was found, and just one C. jejuni isolate from cattle was found to be aminoglycoside resistant, but otherwise sensitive to the other antibiotic classes tested in this study. Aminoglycoside resistance determinants were identified amongst five C. coli STs from AgriFood sources, most commonly from broiler breeder chicken flocks (with multiple isolates taken from a number of flocks) and also 2 sheep and 1 starling.
Multidrug resistance to three or more of the antimicrobial classes tested was rare amongst the human disease isolates from this study, with 13 C. jejuni (0.5% of isolates) being resistant to fluoroquinolones, tetracycline and macrolides. They were most common amongst ST-353CC isolates. Nine C. coli isolates were resistant to fluoroquinolones, tetracycline and aminoglycosides but not macrolides, and one human disease C. coli isolate was resistant to all of the antimicrobial classes tested in this study. Unlike human disease, no Agri-Food isolates were resistant to all of the antimicrobial classes tested in this study.
4. Introduction to the study
4.1. Background
Campylobacter is the most common cause of bacterial gastroenteritis in the UK, resulting in more than double the number of cases annually than all of the other common bacterial foodborne pathogens put together (Daniel et al., 2020). Typically, C. jejuni causes 90% of human disease cases with C. coli causing most of the rest (Cody et al., 2012). There are concerns over the high level of antimicrobial resistance, particularly to fluoroquinolones, and the organism was on the 2017 WHO high priority watch list (Veltcheva et al., 2025). Source attribution models from 25 studies (not restricted to the UK) published between 2001 and 2017 estimate between 60-80% of human campylobacteriosis is derived from chicken meat (Cody et al., 2019; Oxford, 2019; Sheppard et al., 2009). Subsequent concerted efforts have successfully reduced the number of most heavily contaminated UK poultry flocks at slaughter amongst major retailers, but less so amongst smaller and independent retailers (Jorgensen et al., 2019). Similarly, there have been moves to improve antimicrobial stewardship both on farms and in clinical settings, although our previous study found interpretation of the National Institute for Clinical Excellence (NICE) guidelines to vary between regions in practise (Oxford, 2019). Ongoing surveillance for Campylobacter is essential to assess the success or otherwise of interventions such as these.
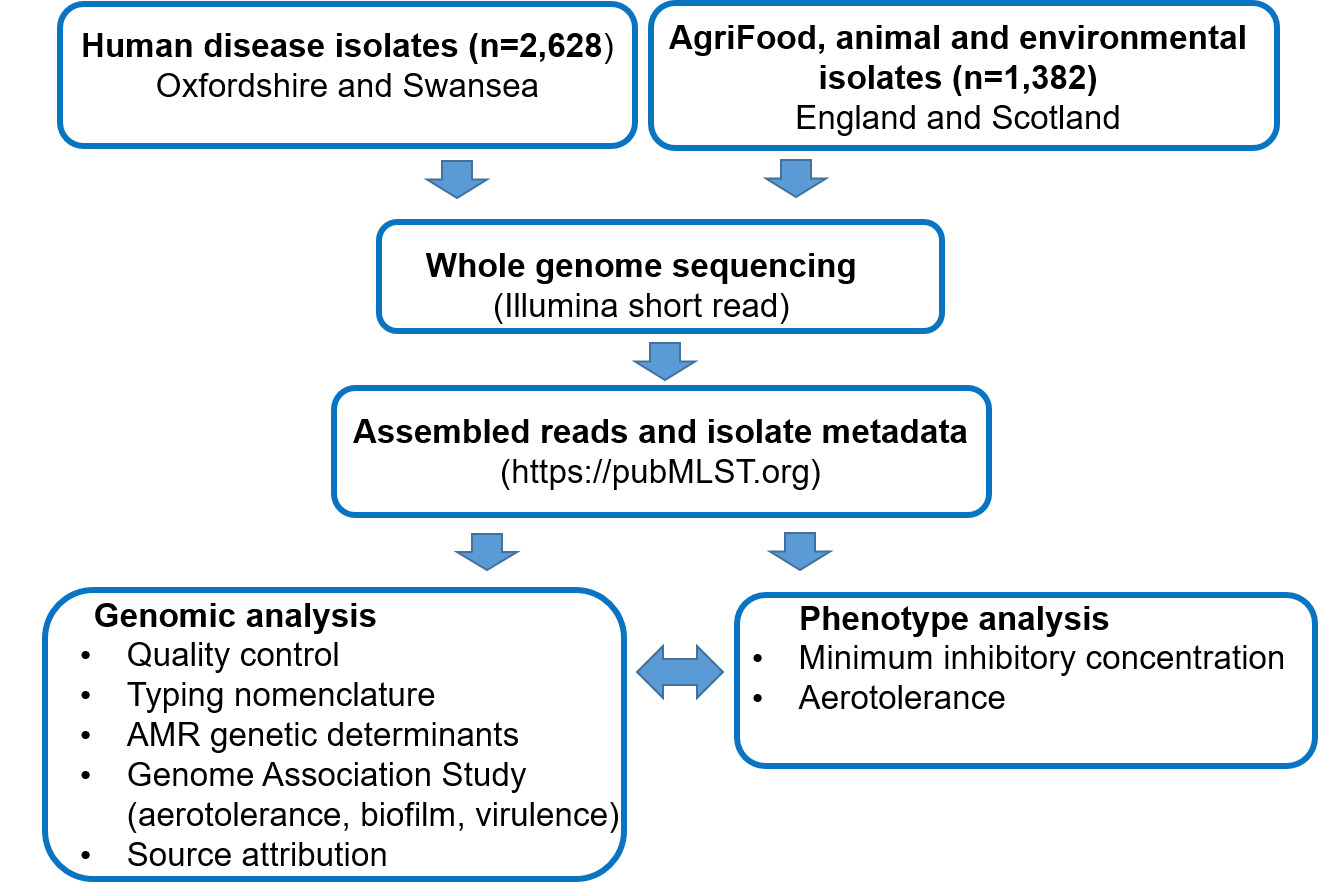
4.2. Aims and overview of the study
With the last UK human disease source attribution study (Oxford, 2019) completed 6 years ago, the aims of this study were to i) determine the current levels of AMR amongst UK human disease and AgriFood isolates and ii) to characterise Campylobacter transmission routes in Agri-Food systems and assess the effectiveness of ongoing interventions. Isolates from previously under sampled sources of infection, including cattle, sheep, deer, horses, extensively reared broiler (meat) chickens and broiler breeder flocks, geese and starlings were included to increase knowledge and accuracy of the source attribution analysis. PubMLST was used to store and analyse the isolate information and DNA sequencing data. Genomic mechanisms underlying AMR were investigated and typing methods for fine-scale epidemiology were enhanced. PubMLST provides the definitive nomenclature for DNA based typing schemes for Campylobacter, enabling further analysis of the impact of globally integrated food distribution systems. Figure 1 shows an overview of the study approach, with methods presented in more detail in section 9 ‘materials and methods’. In brief, Campylobacter isolates from cases of human disease in Oxfordshire (England) and Swansea (Wales) were DNA sequenced, together with contemporary and historical isolate collections from UK AgriFood sources and wild birds. The study used genomic data to predict AMR and virulence determinants for each Campylobacter isolate, as well as to model source attribution. Where possible, phenotyping to determine Epidemiological cut-off values (ECOFFs) on a subset of isolates representing different lineages (clonal complexes) and resistance determinants was performed to validate genomic prediction of AMR. It should be noted that ECOFFs do not necessarily relate to clinical levels of resistance.
4.3. Human Campylobacteriosis
Campylobacter causes approximately 60,000 cases of laboratory confirmed human disease a year in the UK (Graham et al., 2024; UKHSA, 2025). For every confirmed case, there is estimated to be a further 9.3 cases unreported in the community (Tam et al., 2012). The incidence of Campylobacter infection is more than double other common foodborne bacterial pathogens put together (Daniel et al., 2020). Symptoms in previously healthy people most commonly include diarrhoea, vomiting, abdominal pain and fever for up to 7 days (Goddard et al., 2022). Infection is usually self-limiting with antimicrobial treatment reserved for young, elderly or immunocompromised individuals. On rare occasions, severe sequelae such as Guillain Barré Syndrome (GBS), reactive arthritis (ReA) or irritable bowel syndrome (IBS) may develop. There are approximately 21 deaths a year from Campylobacter (Holland et al., 2020). GBS may result in the death of 1 in 20 patients (Shahrizaila et al., 2021).
4.4. Molecular epidemiology of Campylobacter spp
Molecular typing of Campylobacter has been essential to help elucidate the complex biology of the organism. Multilocus sequencing (MLST) using seven loci has been remarkably successful given the relatively low typing resolution. Clustering of closely related sequence types (STs) into clonal complexes (CCs) on the basis of sharing four or more alleles allows convenience for describing lineages with restricted host association, or more generalist colonisation of multiple host sources (Dingle et al., 2002; Sheppard et al., 2010). With the more recent availability of whole genome sequencing data, the MLST approach has been extended to describe variation amongst 1,343 core genes across C. jejuni and C. coli in version 1 of the cgMLST (core genome multilocus typing) scheme (Cody et al., 2017), or indeed, across the entire genome (whole genome multilocus typing, wgMLST). Whole genome sequencing data are backwards compatible with seven locus MLST sequence types. DNA sequencing data and isolate metadata are curated and stored, enabling species identification, comparison of lineages, genes or SNPs of interest such as AMR determinants, and further downstream genomic analyses.
4.5. Antimicrobial resistance
Resistance to the following classes of antimicrobials used commonly in treatment of Campylobacter infection and/or in agriculture were predicted from genetic determinants for each of the isolates. Genomic analysis has been previously shown to predict resistance/sensitivity for these antimicrobial classes with a high degree of confidence, although in this project we seek to confirm the extent of resistance across the diversity of lineages in a population study, and include multiple resistance determinants.
-
Fluoroquinolone resistance.
Fluoroquinolone resistance in C. jejuni and C. coli species is associated with mutations in gyrA, encoding a subunit of the DNA gyrase enzyme (Aleksic et al., 2021). The Thr-86-Ile point mutation, resulting in an amino acid change from Threonine to Isoleucine, is most commonly observed in clinical isolates, resulting in high level fluoroquinolone resistance. Other mutations conferring fluoroquinolone resistance observed to date include single point mutations Ala-70-Thr, Thr-86-Ala, Thr-86-Lys, Asp-90-Asn, and Pro-104-Ser, and double mutations Thr-86-Ile combined with Asp-85-Tyr, Asp-90-Asn, or Pro-104-Ser (Aleksic et al., 2021; Bachoual et al., 2001; McIver et al., 2004; Schiaffino et al., 2024). The Thr-86-Ala point mutation has been associated with resistance to nalidixic acid but low level ciprofloxacin resistance (Bachoual et al., 2001; Dahl et al., 2021; Iovine, 2013). The Thr-86-Val mutation has been identified in C. lari and C. jejuni (Aleksic et al., 2021).
-
Tetracycline resistance.
Tetracycline resistance is associated with the tet(O) gene which is most commonly carried on a plasmid, but can also be found chromosomally on occasions (Iovine, 2013). It was beyond the scope of this project to quantify the carriage of chromosomal versus plasmid encoded resistance determinants. There are important implications however, for example, plasmid or transposon encoded resistance determinants may move more easily between lineages.
-
Macrolide resistance.
Macrolide resistance is generally found to be higher amongst C. coli than C. jejuni isolates (Bolinger & Kathariou, 2017). It is most commonly associated with a A2075G point mutation in the 23S rRNA gene, or less commonly with an A2074C/G point mutation, conferring high-level resistance to macrolides. There is concern over the global spread of the emerging ermB gene (Bolinger & Kathariou, 2017), which confers resistance to macrolides but is also associated with multidrug resistant genomic islands.
-
Aminoglycoside resistance.
Resistance to aminoglycosides is conferred by the presence of genes including ant(6)-Ia, aadE and aadE-Cc (conferring streptomycin resistance), aph(2″)-If and aph(3′)-III (conferring gentamicin resistance) (Dahl et al., 2021; Ortega-Sanz et al., 2025; Schiaffino et al., 2024). Point mutations in rpsL (K43R and K88R) also confer streptomycin resistance.
-
Multidrug resistance.
Campylobacter isolates resistant to 3 or more classes of antimicrobials were defined as being multidrug resistant. In some regions of the world, there is increasing prevalence of a Resistance-Enhancing cme efflux pump which has been associated with multidrug resistance and found to increase resistance to ciprofloxacin 9-fold and erythromycin 4-fold (Schiaffino et al., 2024; Yao et al., 2016).
5. Materials and methods
5.1. Campylobacter isolates
Oxfordshire human Campylobacter isolates were collected from the Oxford University Hospitals NHS Trust clinical microbiology laboratory, which serves approximately 1% of the UK population. The isolates in this study, collected from April 2019 to December 2024, are part of a collection from the Oxford University Hospital Trust’s John Radcliffe Hospital clinical microbiology laboratory that has been ongoing for 22 years, starting in 2003. Previous studies demonstrate that results from this sample site reflect national surveillance (Cody et al., 2012; Oxford, 2019).
Faecal samples that tested positive for Campylobacter by PCR in the clinical laboratory were cultured onto mCCDA. All bacterial growth after 48 hours microaerobic incubation was swept onto a charcoal amies transport swab, enabling transfer to the Oxford University research laboratory in a different location. In the second laboratory, the swabs were cultured onto a fresh mCCDA plate, and presumptive Campylobacter colonies that had characteristic Gram stain, and positive catalase and oxidase reactions were then subcultured onto Columbia Blood Agar to check for purity before DNA sequencing. Campylobacter isolates were frozen in BHI and glycerol broth at -80°C for long term storage. Species identification was based on whole genome sequencing data.
Oxfordshire human disease isolates from this study (2019-2024) were compared with data from isolates collected during earlier years (2003-2018) which can be found by searching for project id 1 ‘Oxfordshire Human Surveillance’ on the PubMLST database isolates tab; Oxfordshire Human Surveillance - Campylobacter jejuni/coli isolates. Note, whole genome sequencing data is not available for years 2005 and 2008 and is less frequent for isolates collected before 2010. Users will need to create a free login account on PubMLST to view all of the isolates.
Human disease isolates from Swansea in Wales were recovered from a collection held in long-term storage at -80°C on beads. They originated from faecal samples submitted to the Singleton Hospital in Swansea, Wales in 2012-2013. Unfortunately, many isolates from the Swansea collection were non-viable following movement between a number of institutions over the years, and consequently most of the downstream analysis in this project focuses on the Oxfordshire collection.
AgriFood, animal and environmental isolates were retrieved from long-term storage collections held by the University of Oxford and from on-going studies by the Moredun Research Institute in Scotland.
The isolate collection for this study can be viewed in a publicly accessible project PATH-SAFE years 1 and 2 combined - Campylobacter jejuni/coli isolates (project id 142).
5.2. DNA extraction and sequencing
For isolates from England and Wales, DNA was extracted using Promega Maxwell® RSC automated DNA extraction instruments and the Promega Maxwell® RSC cultured cells DNA kit (AS1260). DNA was extracted from Scottish isolates using the Qiagen DNeasy Blood and Tissue DNA extraction kit. DNA was quantified using the Promega Quantus fluorometer, normalised to 30 ng/µl in 50µl and submitted for whole genome sequencing using Illumina short-read sequencing technology. Assembled sequences were uploaded onto the PubMLST database together with isolate metadata (https://pubmlst.org). Alleles for each of the isolates were tagged using automated scripts. New alleles were curated and assigned new numbers manually, along with new STs. Campylobacter species was determined from DNA sequence and ribosomal MLST (rMLST) profile (Jolley et al., 2012).
5.3. Development of the PubMLST database.
5.3.1. Molecular typing schemes
PubMLST (https://pubmlst.org) is a publicly available web accessible database that stores and catalogues isolate metadata and whole genome sequencing data and integrates a variety of tools for downstream analysis (Jolley et al., 2018). The internationally adopted genomic nomenclature provided by PubMLST is essential for molecular surveillance of Campylobacter and underpins research on food-borne pathogens forming part of the PATH-SAFE project, as well as other microbes.
As part of this project, the PubMLST database capabilities have been enhanced to improve detection and reporting of AMR genetic determinants. These include detection of single nucleotide polymorphisms (SNPs) within DNA nucleotide sequence, or amino acid changes and gene presence/absence. Antimicrobial class specific schemes have been created, allowing the user to search for all genes and their allelic variants at the same time. We have similarly created schemes for genes associated with virulence, aerotolerance and biofilm formation. These changes are fully integrated into BIGSdb (Jolley et al., 2018) (the software underlying PubMLST) allowing querying, export, and analysis via the web interface. For example, the entire global collection of more than 150,000 Campylobacter isolates can be searched for the presence of the Thr-86-Ile point mutation causing fluoroquinolone resistance within 5 seconds at the click of a button. Access to scheme definitions is available via the application programming interface.
We have enhanced the web-based genome annotation tools within BIGSdb to automatically flag sequences with ambiguous bases that would otherwise be complete coding sequences, and to mark alleles that are incomplete due to being broken by contig ends at user-defined identity thresholds. In addition, stop codons are automatically flagged and colour coded to indicate their position within a locus. The changes allow significant improvements in the time required to curate data submissions.
5.3.2. Core Genome Multilocus Sequence Typing scheme version 2 (cgMLST v2) for Campylobacter jejuni/coli.
The first core genome MLST scheme for Campylobacter jejuni/coli was published in 2017 (Cody et al., 2017). Since then there have been many thousands more Campylobacter isolates sequenced and we have updated the cgMLST scheme to version 2, to more fully encompass the global diversity that was not available at the time the original scheme was developed. The additional isolates have enabled us to review problematic loci with respect to issues such as alternative, inconsistent or multiple start sites, internal stop codons, phase variation, or loci that are frequently incomplete.
The cgMLST v2 scheme has a total of 1,142 loci, compared to 1,343 loci in v1 of the scheme. Removal of 201 loci has resulted in a more robust scheme allowing cgSTs to be assigned for any isolate with 25 or fewer missing loci (compared to 50 missing loci previously) – we’re now able to assign and cluster cgSTs for 99% of isolates with assemblies containing 100 or fewer contigs.
5.3.3. Cluster detection
As part of a suite of tools for genomic analyses, we have developed a BIGSdb plugin for ReporTree to facilitate automated cluster detection and display (Mixao et al., 2023). Clusters of isolates can be detected at different threshold levels using the cgMLST v2 scheme, generating files for distance matrices, tree drawing, sequence alignment and other down-stream analyses.
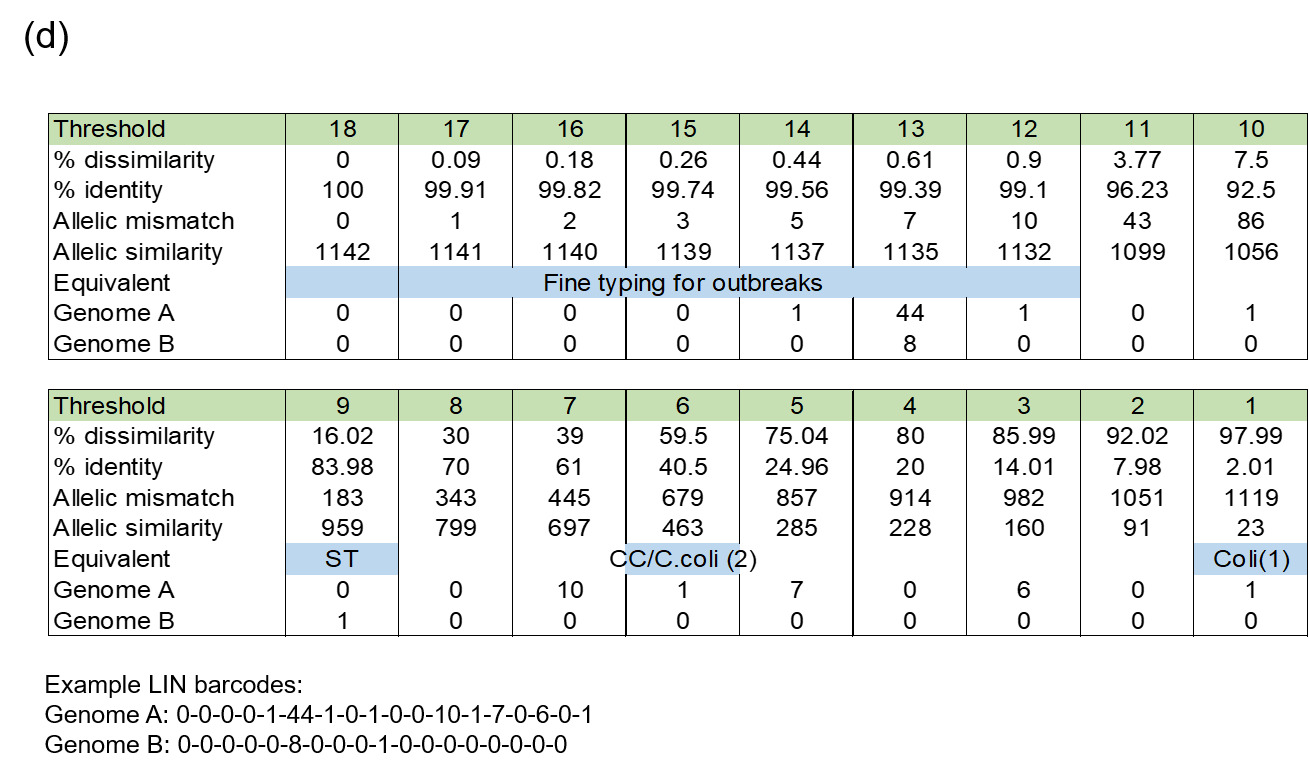
5.3.4. Life Identification number (LIN®) barcoding system
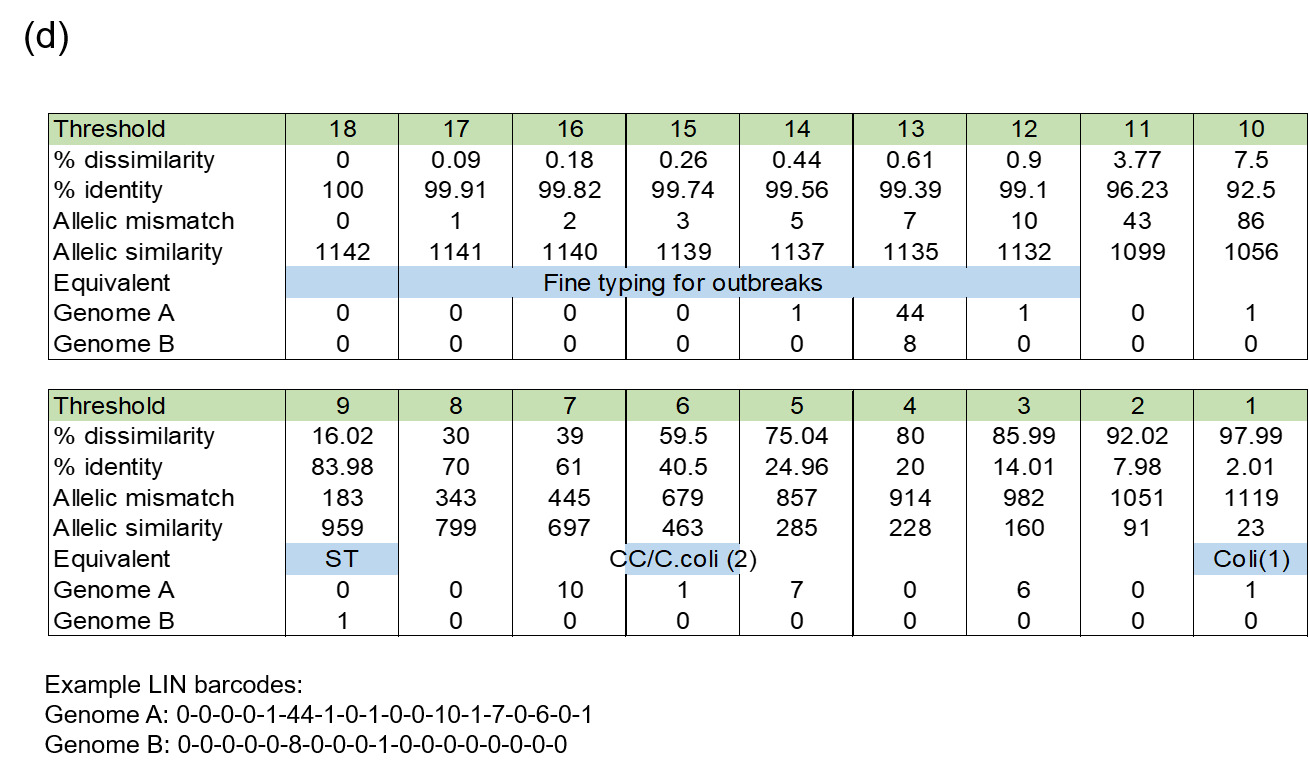
Whilst cgMLST threshold levels are useful for fine-typing isolates at a moment in time, clustering algorithms will need to be repeated for longitudinal analysis, meaning that named clusters are not stable. There can also be a chaining effect if cgSTs are intermediate between different cluster thresholds. A solution to this problem is to use a LIN® bar coding system (trademark registered by This Genomic Life, Inc, Floyd, VA, USA), based upon cgMLST, but where isolates are assigned a profile identification that will not change (https://bigsdb.pasteur.fr/klebsiella/cgmlst-lincodes/). A stable nomenclature is particularly important for investigating the epidemiology of Campylobacter outbreaks which are most often diffuse in nature, in contrast to more classical outbreaks seen with other organisms such as Salmonella.
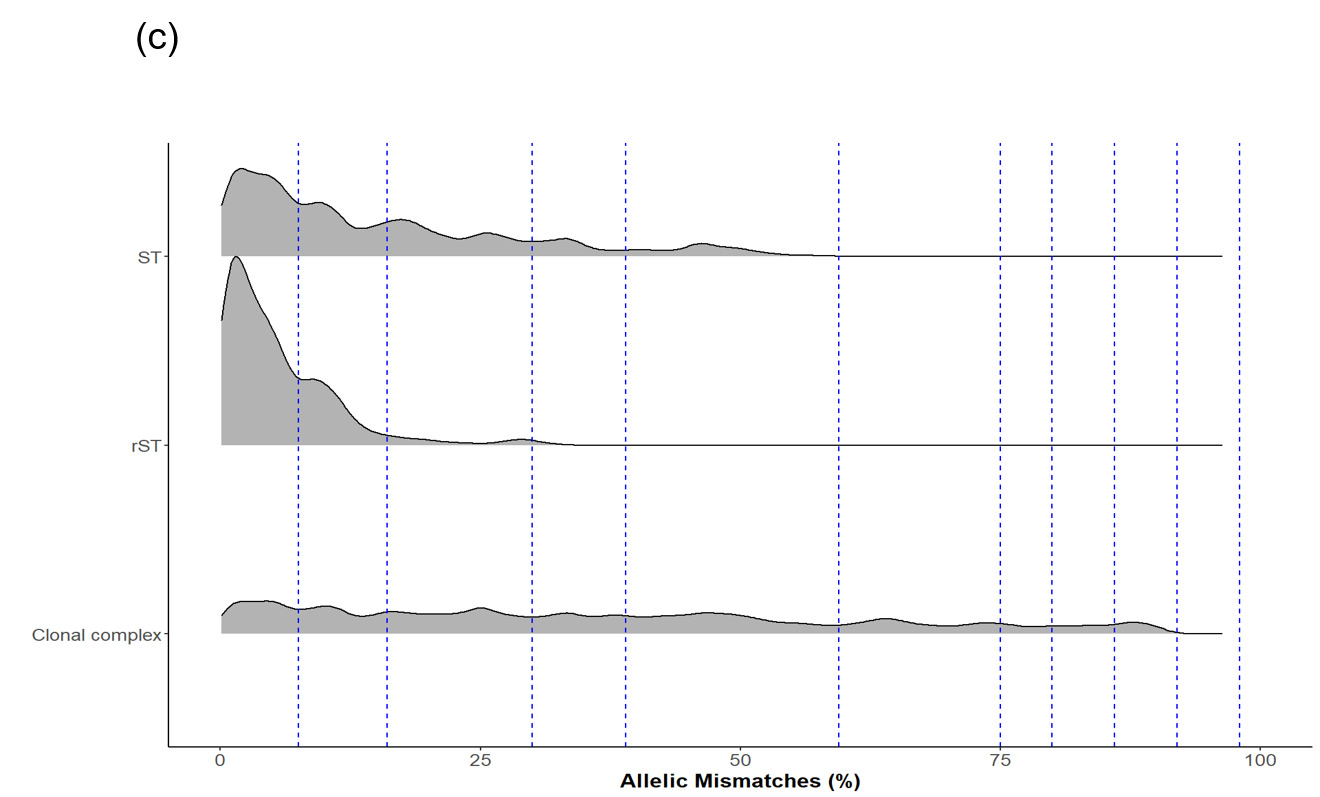
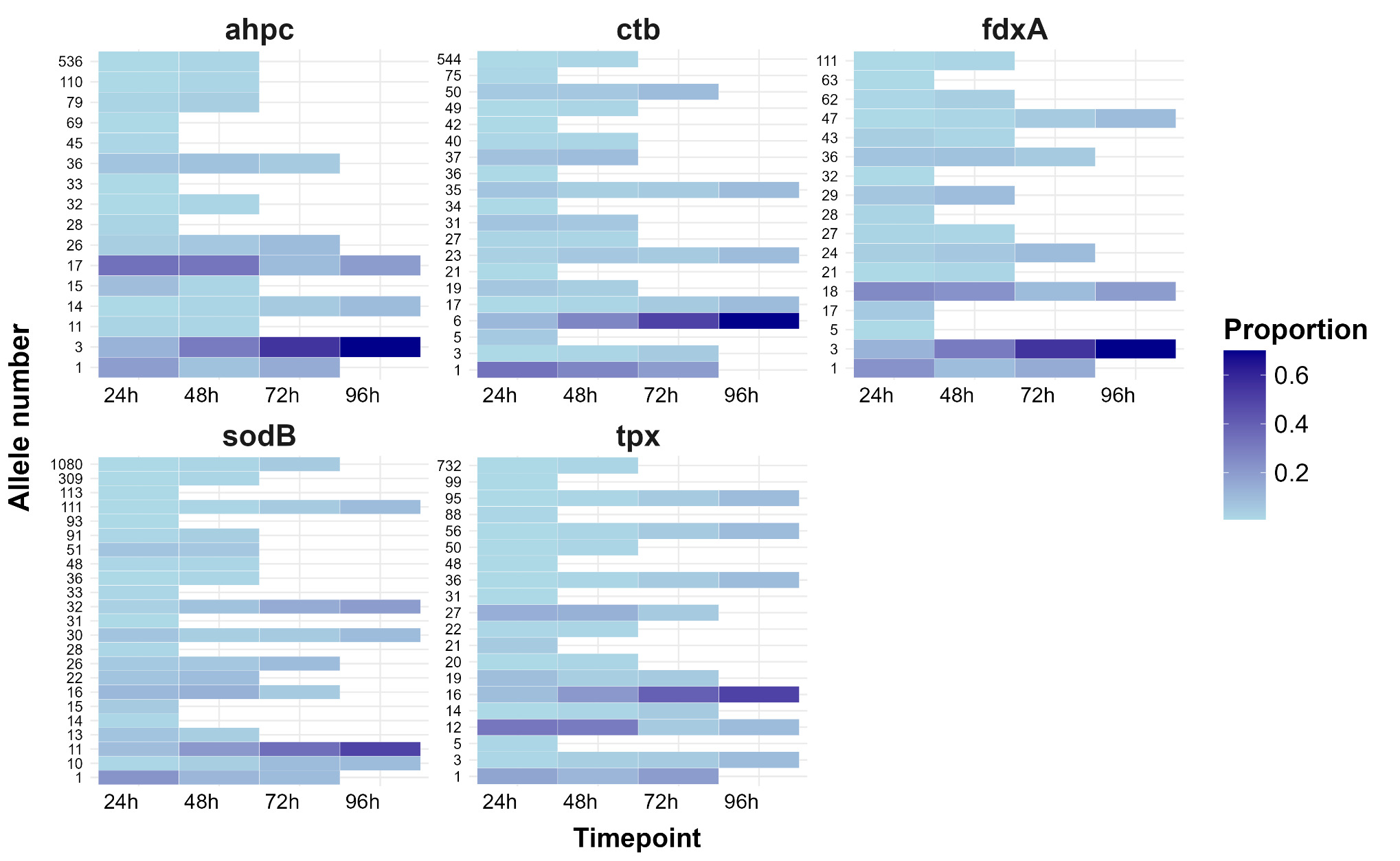
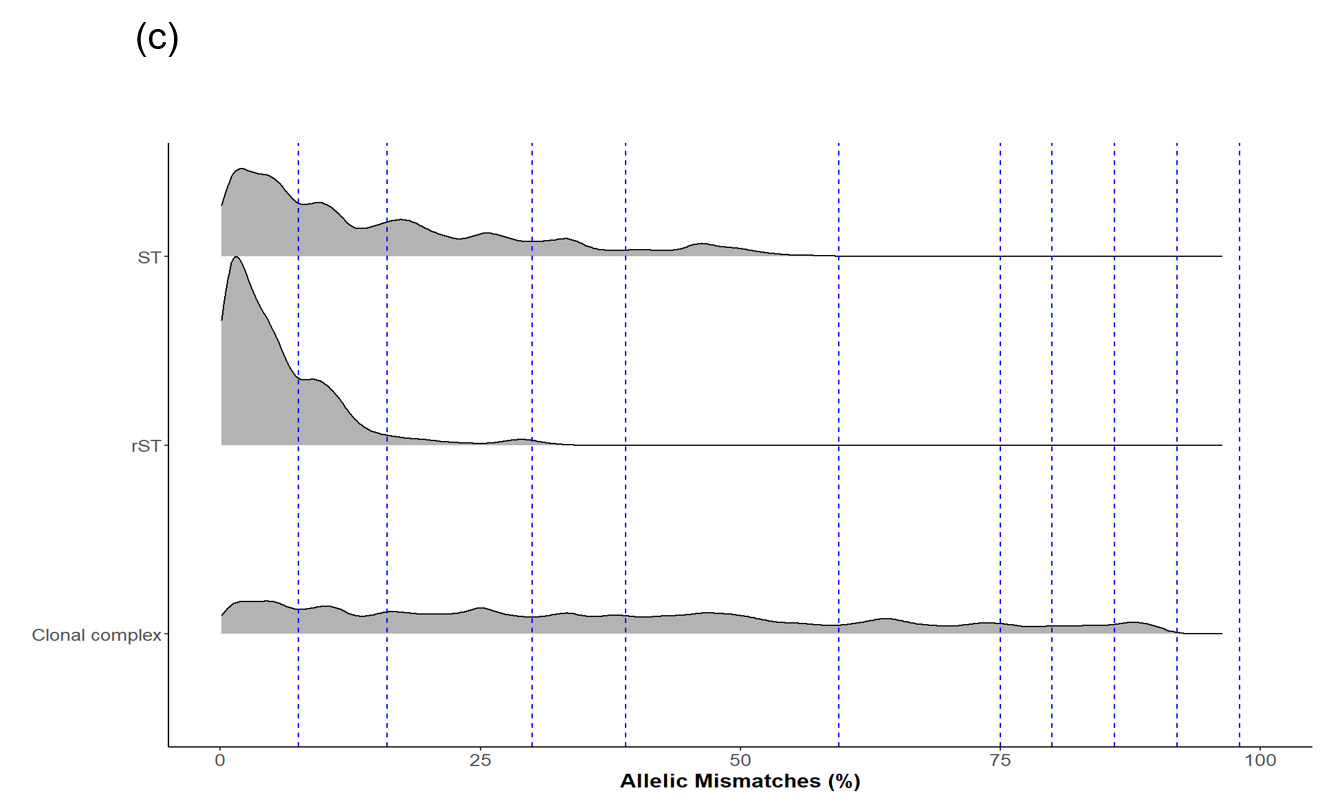
The LIN code system for Campylobacter (Parfitt et al., 2026) was developed using the same methodology as other pathogens including Klebsiella pneumoniae (Hennart et al., 2022), Streptococcus pneumonia (Jansen van Rensburg et al., 2024), Neisseria gonorrhoea (Unitt et al., 2025) and Neisseria meningitidis (unpublished). However, unlike these organisms, we have included two species, both C. jejuni and C. coli, in the scheme due to the high level of introgression of C. jejuni alleles by C. coli (Sheppard et al., 2013). The scheme also includes ancestral C. coli (Sheppard et al., 2008) which give the first C. coli split (Figure 2d), whilst the two C. coli clonal complexes ST-828CC and ST-1150CC split at the same threshold as the C. jejuni complexes ('CC/C. coli (2)').
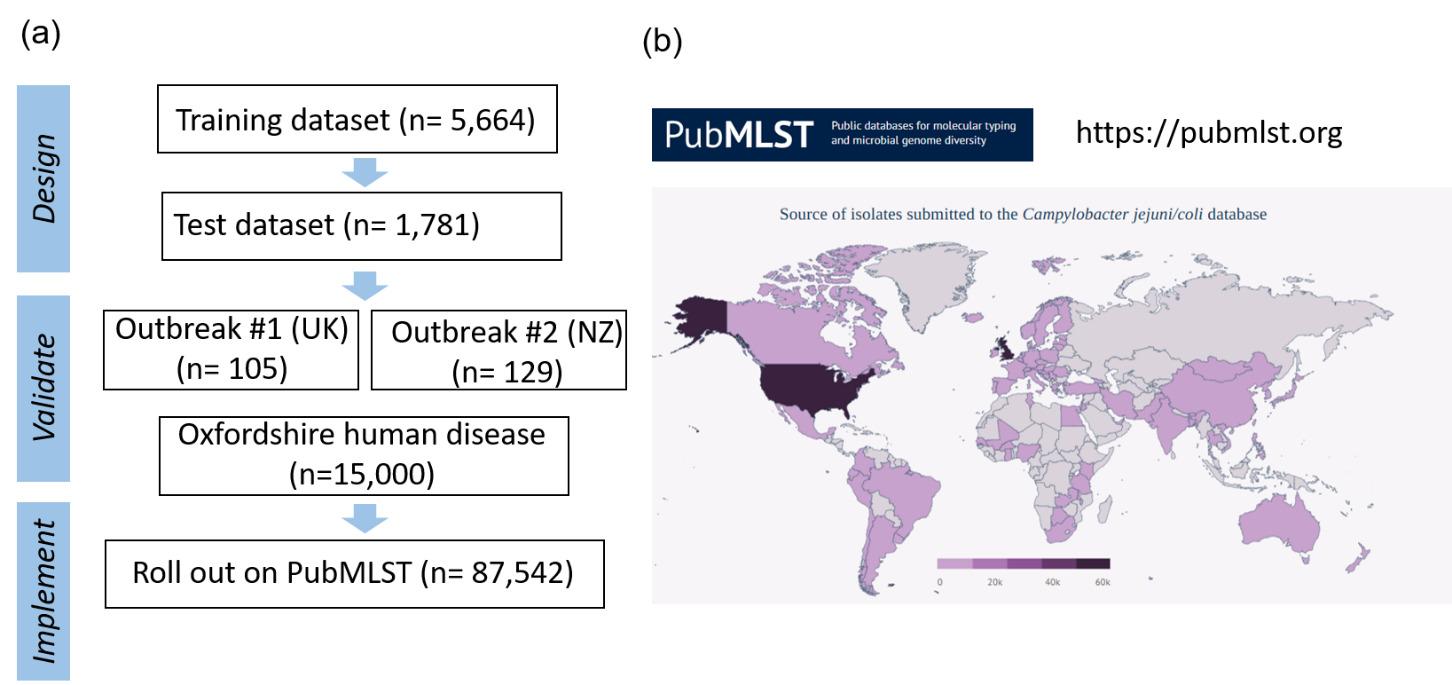
Whole genome sequencing data for C. jejuni and C. coli isolates from the PubMLST database were filtered for quality and then randomly selected using dplyr in R to obtain up to 200 representative isolates per clonal complex. This gave a training dataset of 5,664 isolates (Figure 2). MSTclust (Minimum Spanning Tree (MST)-based clustering) was used to draw a pairwise distance matrix and for statistical analysis. Ridgeline plots were drawn using the ggplot2 package in R, and thresholds were applied where there were natural breaks (peaks and troughs) in the population structure (Figure 2c). The LINcoding Python script (https://gitlab.pasteur.fr/BEBP/LINcoding) was used to assign LIN codes to the isolates, which were then validated using the Grapetree plugin on PubMLST to assess compatibility with existing nomenclature systems such as 7-locus MLST, cgMLST v2 and ribosomal MLST (rMLST) (Jolley et al., 2012). Note, LIN barcode profiles are designed to be computer rather than human readable and existing nomenclature gives biological relevance and context that is more easily conveyed in conversation.
The LIN codes were further validated using two published Campylobacter outbreak datasets (Fernandes et al., 2015; Gilpin et al., 2020), and the Oxfordshire human disease dataset from this study, before being rolled out across more than 99,000 Campylobacter isolates with whole genome data on PubMLST.
_*campylobacter*_training__test_and_validation_sets_that_were_sourced_from_(b.png)
5.3.5. AMR resistance determinants; comparison of PubMLST with other AMR search platforms using ABRicate.
Using a subset of 4,477 Oxfordshire human disease C. jejuni isolates spanning the years 1997-2018, ABRicate (https://github.com/tseemann/abricate) was used to screen the CARD (Jia et al., 2017) and ResFinder (Zankari et al., 2012) databases for key AMR genes predicting resistance to tetracycline, aminoglycosides and macrolides (ermB gene) (Table 1). It should be noted that there is variation in the number of resistance determinants and nomenclature between all of the databases, and here, the comparison between them is to give an overview rather than critical assessment. It was not possible to screen for point mutations conferring resistance to fluoroquinolones or macrolides using ABRicate (though this can be done using the databases individually). Only full coding sequences for genes have been included in the counts for PubMLST. Resistance determinates with partial or truncated coding sequences, or sequences with internal stop codons were found in additional isolates, but it cannot be certain if the genes relate to a functional phenotype.
PubMLST showed good agreement with the other databases tested in this study, predicting tetracycline resistance for 75.2% of the test isolates compared to 75.6% for both CARD and ResFinder. Similarly, aminoglycoside resistance was predicted for 2.7% of the test isolates using PubMLST compared to 3.1% for CARD and ResFinder. The small differences between the prediction for tetracycline and aminoglycoside resistance most commonly result from truncated genes in the sequencing assemblies, and it should be noted that the resistance levels reported in this study using PubMLST are a conservative prediction. AMR phenotype showed good concordance with genotype prediction by PubMLST however.
5.4. Interactive Dashboard
All genome and isolate meta data from the study can be explored and analysed using the PubMLST database. However, these data are complex, multi-dimensional and challenging to comprehend in a spreadsheet or similar format. We transformed these inherently non-visual data into intuitive visual formats to support visual analysis and understanding of these data. We developed two integrated web-based dashboards for the exploration of i) the human disease isolates and ii) the AgriFood, animal and environmental isolates which can be accessed here; Campylobacter Dashboard. Users can interactively visualise and explore the data to detect trends, patterns or anomalies in AMR over time thus gaining insight of the situation to facilitate informed decision making.
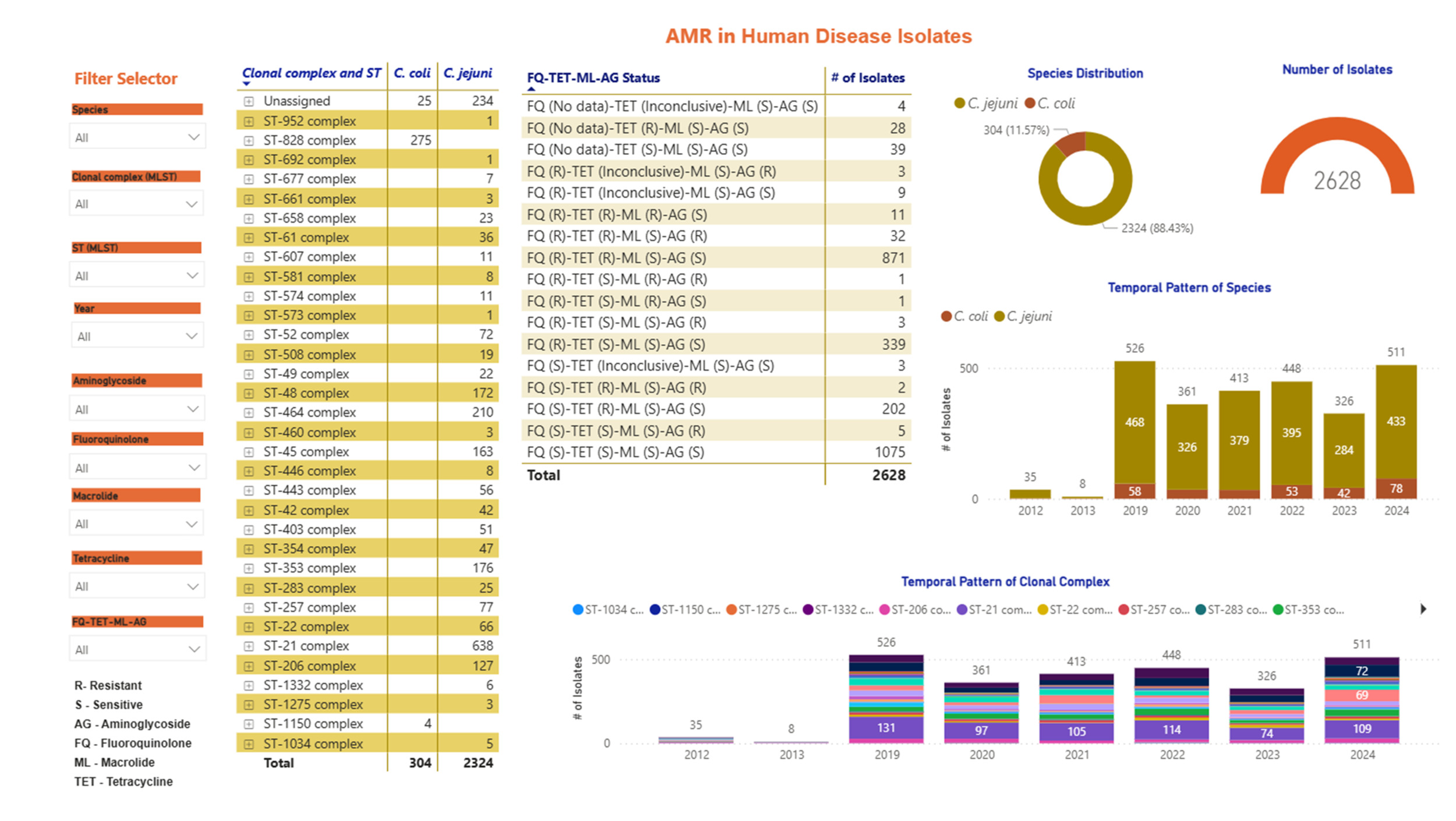
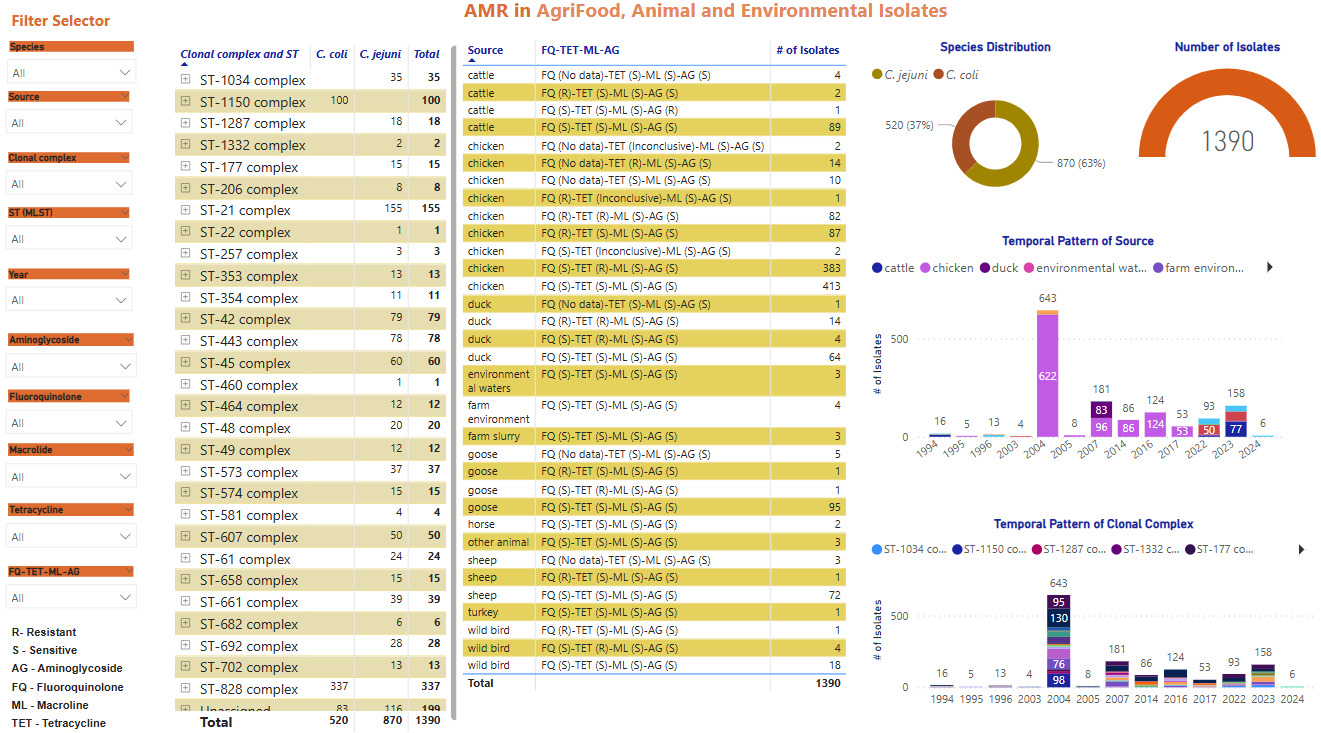
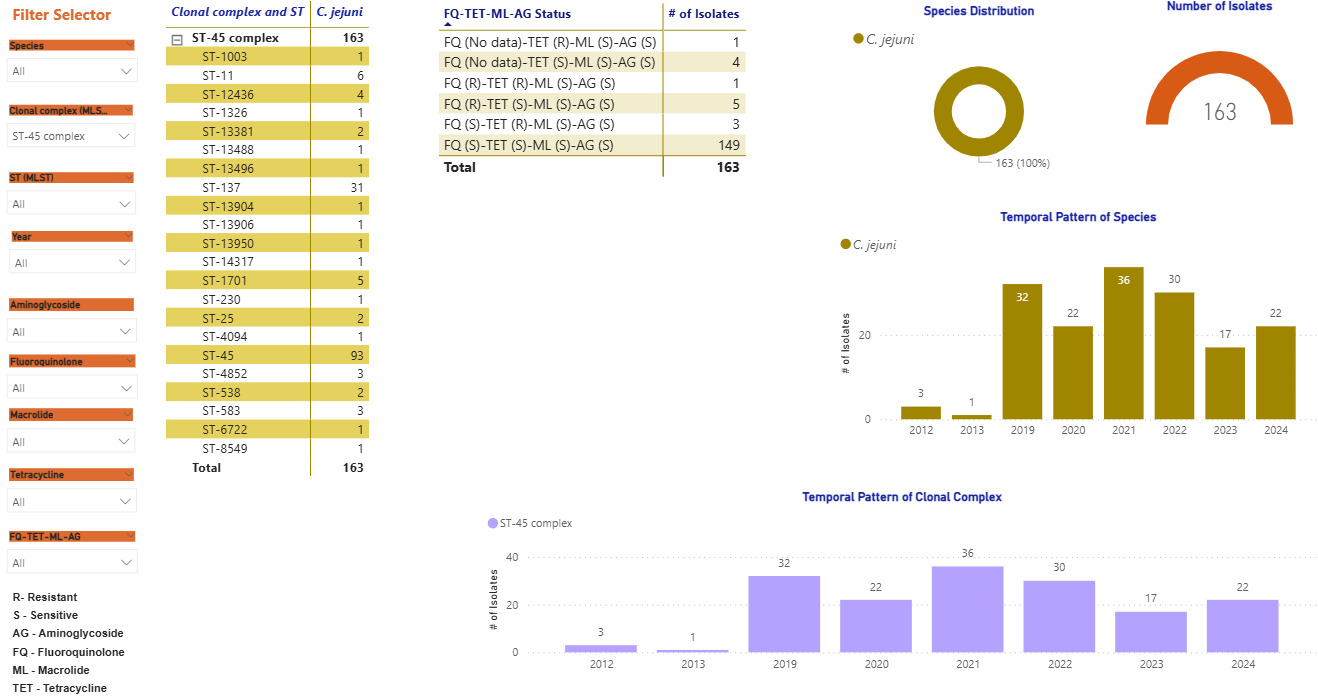
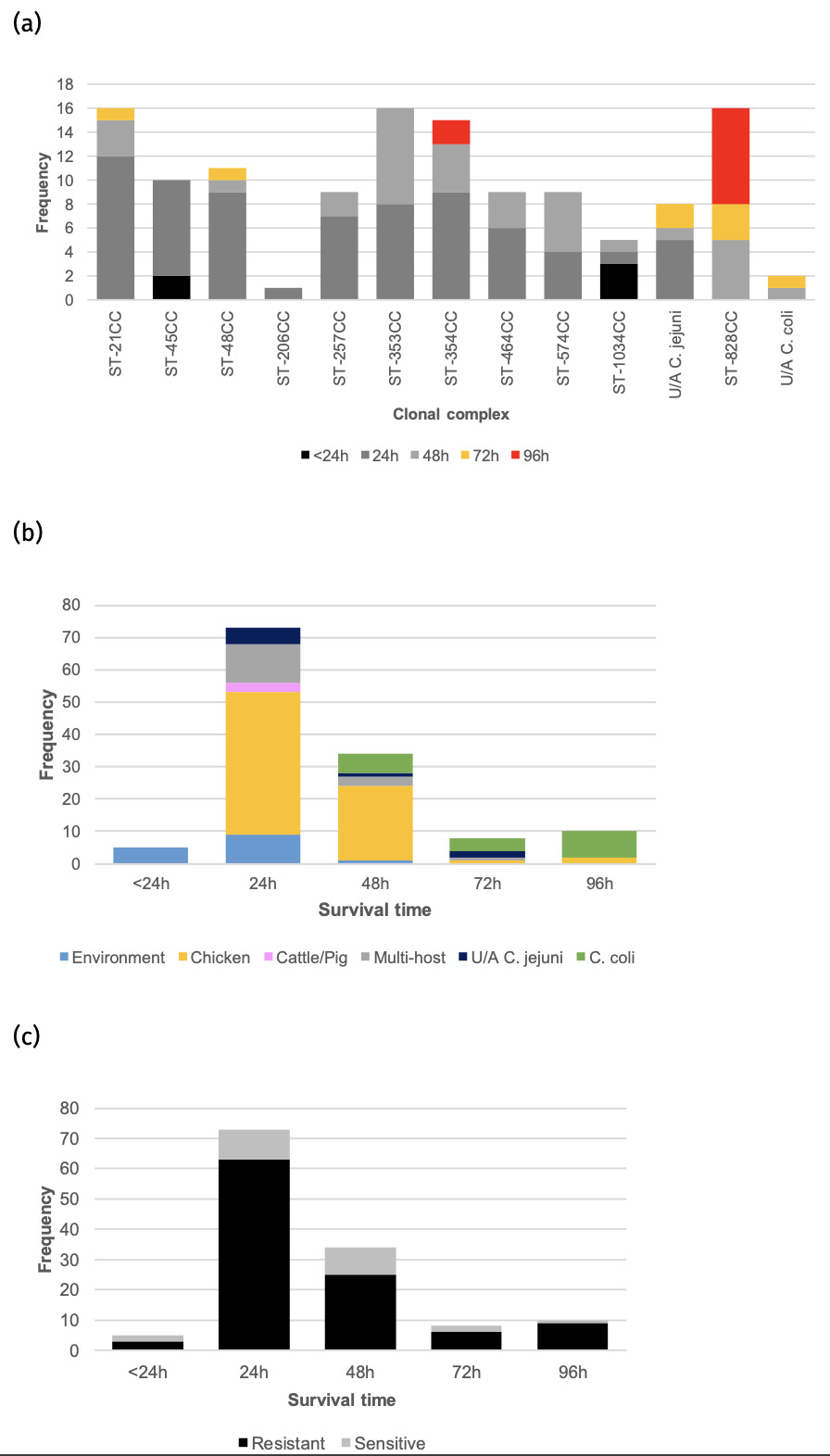
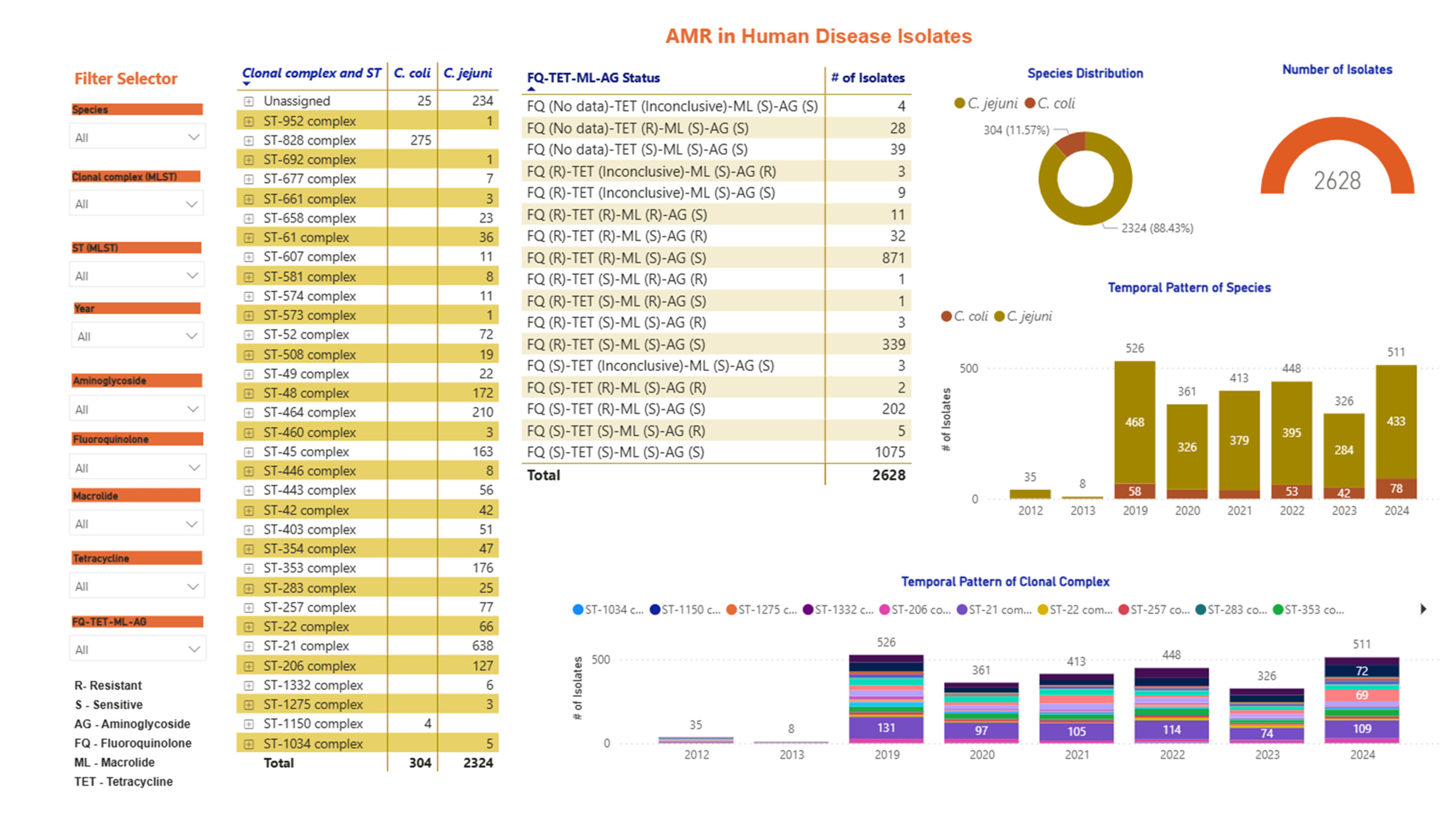
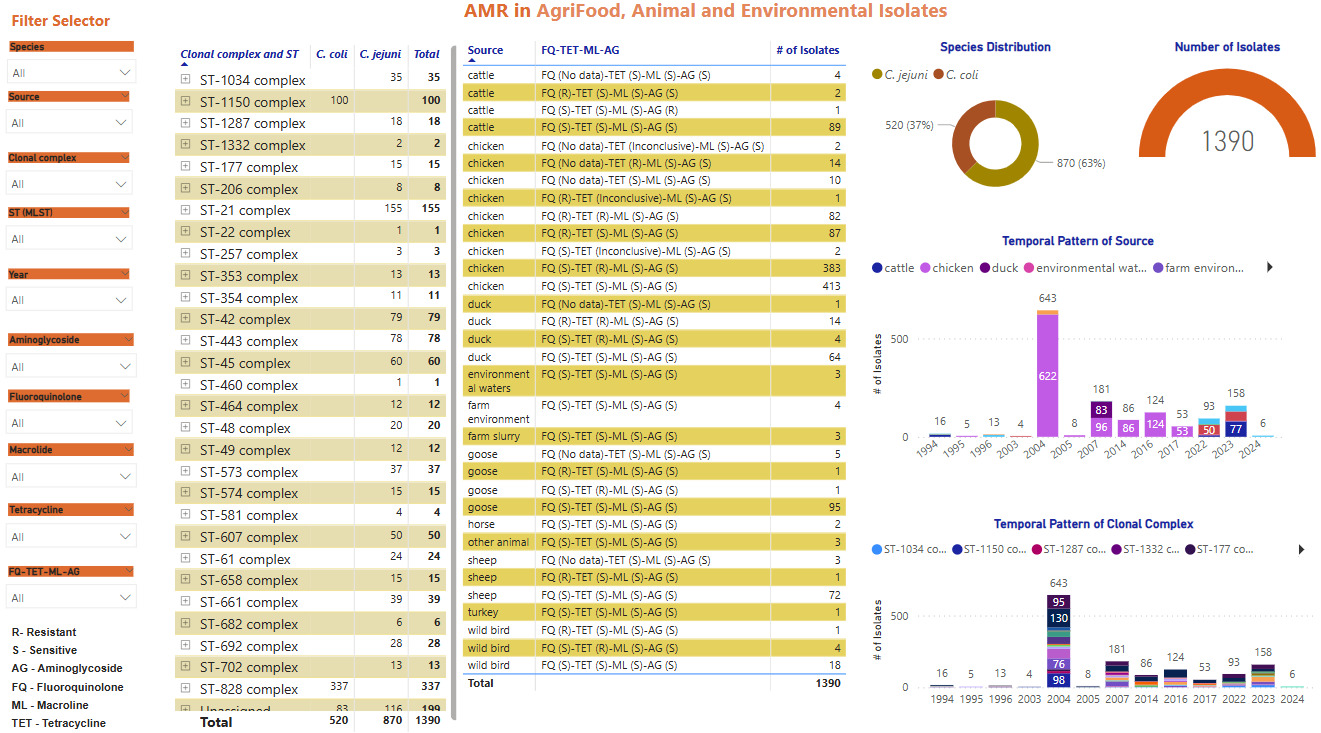
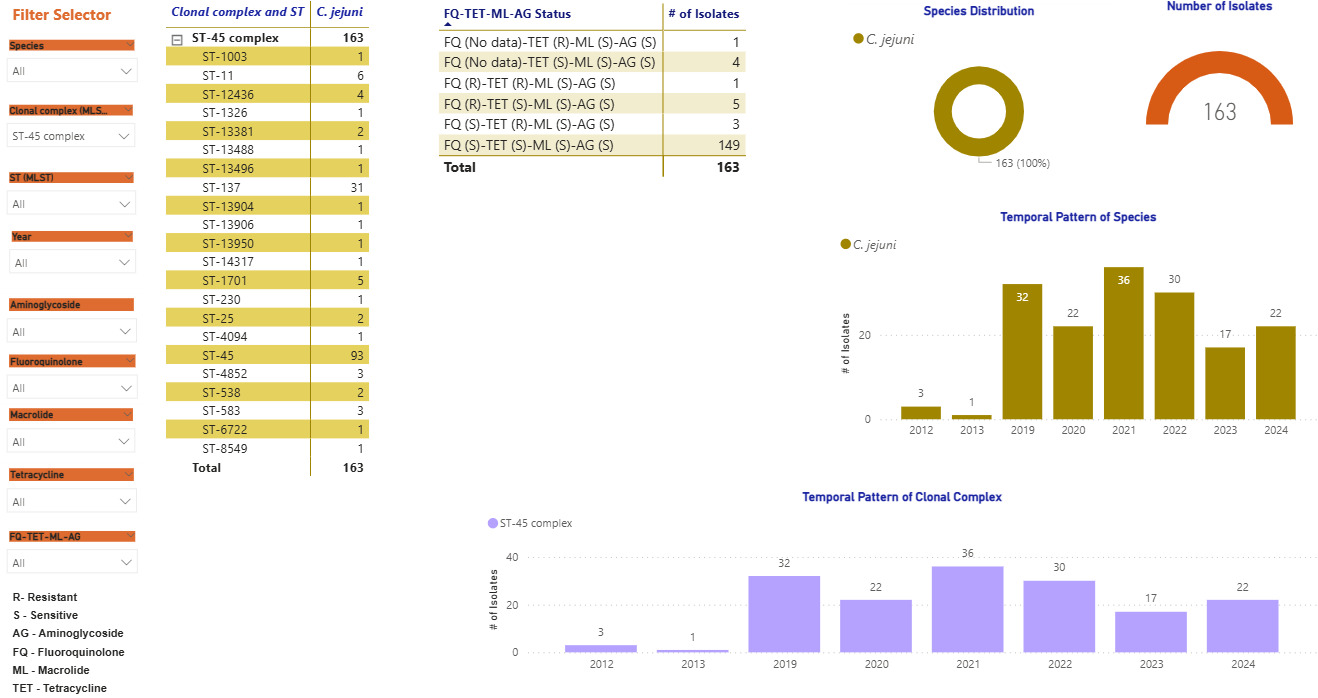
Screenshots of the Campylobacter dashboards are shown in Figure 3 with the format the same for each. In brief, Figure 3a displays an overview of data for human isolates and Figure 3b displays an overview of data for the AgriFood and environmental isolates. Figure 3c gives an expanded view of ST-45 clonal complex isolates amongst human disease.
On the left side, nine drop-down boxes allow specific feature(s) of interest to be filtered in any combination, namely: species, clonal complex, sequence type (ST), year, and sensitivity/resistance to aminoglycosides (AG), fluoroquinolones (FQ), macrolides (ML), tetracyclines (TET), or resistance patterns to each of the antimicrobial classes in combination. The next panel across shows the breakdown and frequency of the filtered isolates into clonal complexes, which may be expanded to show finer ST information by clicking on the plus sign on the left-hand side of each row. The third panel summarises the FQ-TET-ML-AG status and shows the number of isolates with a particular pattern of resistance/sensitivity across all four antibiotic classes concurrently. The remaining panels show breakdown of the selected isolates by species (C. jejuni vs C. coli), source and year. Clicking on any element of the data displayed on the dashboard allows the user to examine the data in greater detail.
Figure 3c gives an example of more detailed exploration of the human disease isolate collection, showing ST-45 and ST-137 to be the most commonly identified STs within the ST-45 clonal complex. The majority of ST-45 isolates were predicted to be sensitive to each of the antimicrobial classes tested, and only small fluctuations in prevalence were seen by year (2012-2013 Swansea isolates, 2019-2024 Oxfordshire isolates).
6. Results
6.1. Human disease isolates.
6.1.1. Oxfordshire human disease isolates
There was seasonal variation in the number of samples testing positive for Campylobacter, with the peak of infection occurring between May and September each year. The number of Campylobacter infections was exceptionally high in June 2021 (133/549, 24.2% of the yearly infection) and samples were diverted to a nationwide UK Health Security Agency study (Swift et al., 2025). The second highest peak was seen in June 2024, accounting for 117/834, 14.0% of infections that year. The number of PCR positive samples was lower in April and September 2020, likely a result of the COVID-19 related lockdowns and associated change in healthcare seeking behaviour (Ondrikova et al., 2021). The number of PCR positive reported Campylobacter infections in Oxfordshire has been increasing year on year since 2021, with a record number seen in 2024, representing more than an 11% increase compared to 2023. A total of 2,968 Campylobacter isolates from April 2019 and December 2024 were submitted for whole genome sequencing (Figure 4).

Table 2 shows the number of Campylobacter isolates recovered from the PCR positive stool samples, together with MLST typing profiles derived from whole genome sequencing data. A number of Campylobacter isolates could not be recovered upon culture from the PCR positive stool samples due to a lower sensitivity of detection, or overgrowth with other bacteria.
6.1.2. MLST typing, all human disease isolates
Seven-locus MLST profiles were identified for a total of 2,628 Campylobacter human disease isolates from Swansea (Wales) and Oxfordshire (England) (Table 2), comprising 2,324 (88.4%) C. jejuni and 305 (11.6%) C. coli isolates (Table 3).
A total of 287 C. jejuni STs and 75 C. coli STs were recovered, grouping into 31 C. jejuni and 2 C. coli clonal complexes respectively (Table 4).
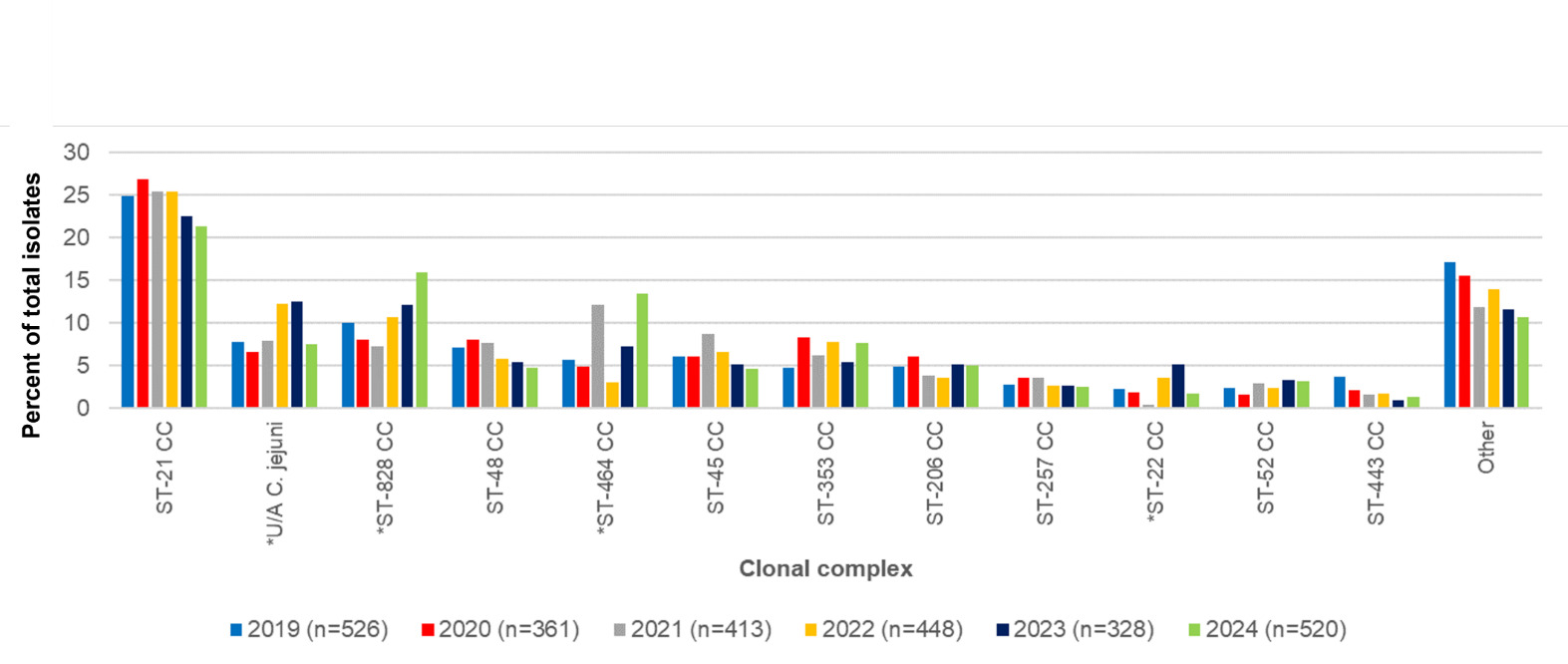
The 5 most frequent C. jejuni clonal complexes for Oxfordshire isolates in years 2019-2024 combined were ST-21CC (n=638/2628, 24.3%), ST-464CC (n=210/2628, 8.0%), ST-353CC (n=176/2628, 6.7%), ST-48CC (n=172/2628, 6.5%) and ST-45CC (n=162/2628, n=6.2%) (Table 4, Figure 5). The C. coli clonal complexes ST-828CC and ST-1150CC accounted for 275/2628, 10.5% of isolates, and 4/2628, 0.2% of isolates respectively. Isolates unassigned to a clonal complex comprised 235/2628 C. jejuni (8.9%) and 25/2628 C. coli (1.0%).
The overall distribution of clonal complexes differed significantly by year (χ2 297.49, 170 d.o.f, p <0.001). Individual C. jejuni clonal complexes that differed significantly by year were ST-464 and ST-22 complexes (Table 5). Additionally, the number of C. jejuni isolates that were not assigned to a clonal complex varied significantly across the years 2019-2024. The C. coli ST-828CC also showed significant difference in prevalence across different years.
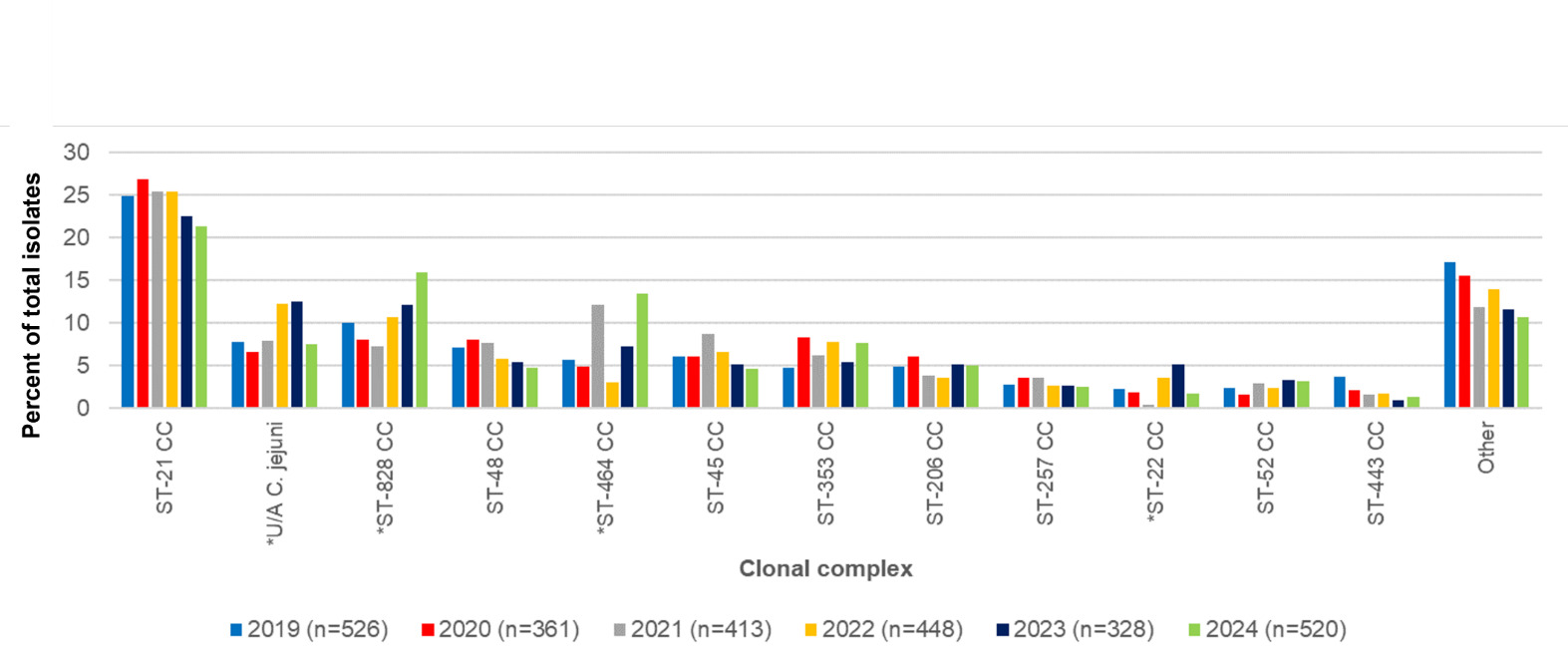
ST-21CC was the most commonly isolated clonal complex isolated every year in the study (Table 6). Other clonal complexes were consistently within the top ten, but changed rankings slightly from year to year. Environment and wild bird associated complexes (eg ST-1034CC, ST-692CC and ST-1332CC) were the lowest ranking in the years they were present and were missing in other years.
The five most common C. jejuni STs recovered from Oxfordshire human disease isolates in the years 2019-2024 combined were ST-6175 (ST-21CC) (n=178/2628, 6.8%), ST-48 (ST-48CC) (n=152/2628, 5.8%), ST-5136 (ST-464CC) (n=149/2628, 5.7%), ST-50 (ST-21CC) (n=127/2628, 4.8%) and ST-21 (ST-21CC) (n=108/2628, 4.1%) (Figure 6). ST-827 (n=60/2628, 2.3%) ST-825 (37/2628, 1.4%), ST-9012 (n=21/2628, 0.8%), ST-829 (n=21/2628, 0.8%) and ST-1055 (16/2628, 0.6%) were the most common C. coli STs. Some clonal complexes were more diverse in terms of STs (eg ST-21CC) compared to others (eg ST-48CC)

6.2. AgriFood, animal and environmental isolates.
6.2.1. Isolate collection
Campylobacter isolates obtained from UK sources under-represented on PubMLST and in national culture collections comprised of cattle, sheep, deer, horses, free-range broiler breeder (meat chicken parent flock), pet/back-yard chicken flocks, turkey, farmed ducks, wild geese and starlings, slurry and environmental water samples (Table 7). The poultry isolates are from rare sample collections including extensively reared broiler (meat) chicken flocks and broiler breeder flocks which were included to explore nuances within this source of infection.
6.2.2. MLST typing
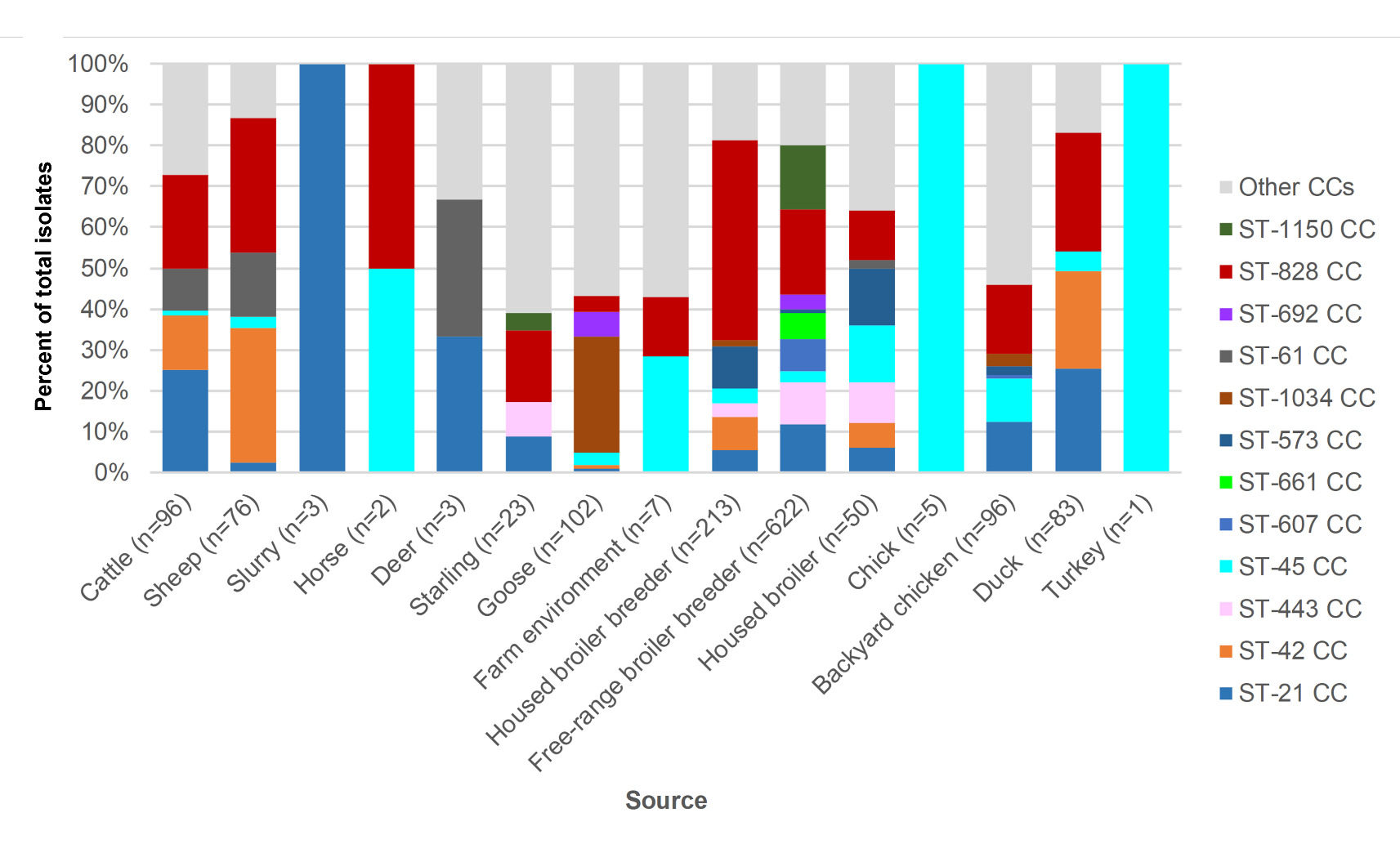
Seven-locus MLST profiles were obtained for 1,382 AgriFood, animal and environmental Campylobacter isolates, of which 870/1382 (63.0%) were C. jejuni and 520/1382 (37.6%) were C. coli (Table 8, Table 9). The C. jejuni isolates are grouped into 27 clonal complexes and the C. coli into the only two clonal complexes ST-828 and ST-1150 complexes. A total of 116/1382 (8.39%) C. jejuni and 83/1382 (6.01%) C. coli isolates remaining unassigned.
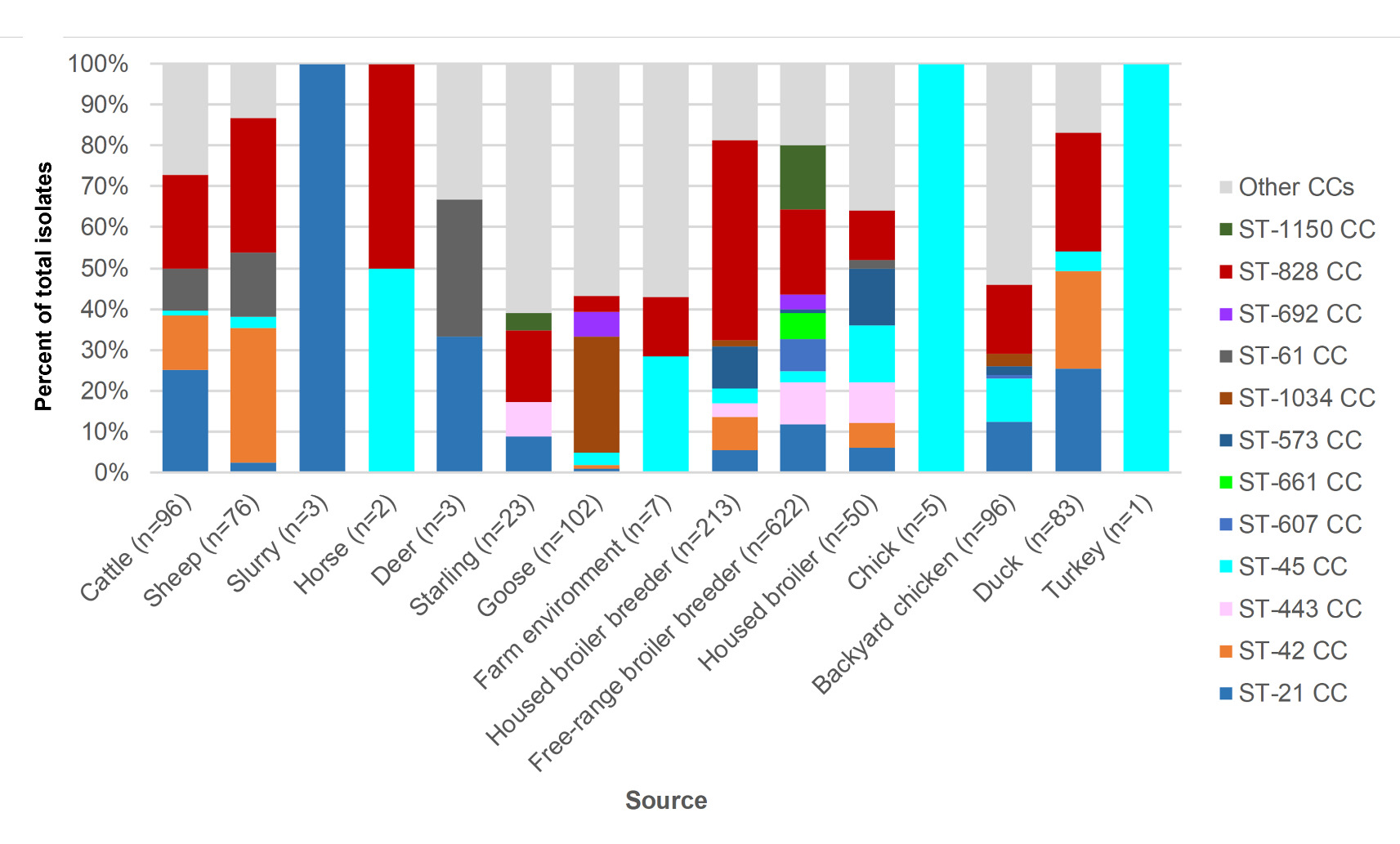
Amongst livestock, wild animal and environmental samples (Table 8), the following C. jejuni clonal complexes were most common; ST-42CC (39/312, 12.5%), ST-21CC (33/312, 10.6%), ST-1034CC (29/312, 9.29%), ST-61CC (23/312, 7.4%) and ST-48CC (15/312, 4.8%). Amongst poultry isolates (Table 9) the following complexes were most common, though the bias towards free-range broiler breeder isolates should be noted; ST-828CC (280/1070, 26.2%), ST-21CC (122/1070, 11.4%), ST-1150CC (99/1070, 9.3%), ST-443CC (76/1070, 7.1%) and ST-45CC (51/1070, 4.8%).
The breakdown of clonal complex by source is shown in Figure 7.
6.2.3. Campylobacter in horses
6.2.3.1. Introduction
In common with other mammals and livestock, healthy horses are thought to carry Campylobacter in their intestines and have been implicated as a source of human infection in a large waterborne outbreak of campylobacteriosis in Norway (Paruch et al., 2020). There have been few studies of Campylobacter in horses, and isolates are very rare, with none recorded on the PubMLST database before this study. Campylobacter is occasionally isolated from sick horses, whilst prevalence of Campylobacter carried by healthy horses ranges from 0-26% using a combination of culture and PCR, with most studies sampling less than 100 individuals (Blunden et al., 2006; Bolton et al., 2012; Browning et al., 1991; Hurcombe et al., 2009; Moriarty et al., 2015; Selwet, 2021; Stout et al., 2021). A study of 304 foals, the majority coming from foals with diarrhoea, recorded just one Campylobacter positive sample (Browning et al., 1991).
No genotyping information exists for Campylobacter from horses. The Norwegian waterborne outbreak attributed riding horses from a nearby stables to be the source of infection using host specific bacteriodales 16S rRNA markers, rather than source attribution analyses using Campylobacter typing information (Paruch et al., 2020).
Within the time and resources available, we set out to isolate Campylobacter from healthy riding horses in the Oxfordshire region, and compare genotyping data with two isolates we had in our freezer archives from health screens of adult horses that were hospitalised in a veterinary clinic.
6.2.3.2. Methods
Fresh faecal samples were collected from 33 horses (43 samples), from which up to 11 samples might be expected to be culture positive based upon previous published studies. The horses were sampled from two different farms and also at a weekend riding training camp where horses originated from a variety of locations across Oxfordshire. The ages of the horses ranged from 6 to 27 years and they were a variety of breeds (figures 8 and 9). Samples were collected in June and July when a summer peak of infection might be expected. Horses on farm 1 were on grazing that had been flooded over the winter, and neighbours a bird reserve. Ten of the horses from farm 1 were sampled on 2 occasions, 1 month apart.
The faecal samples collected from healthy horses were also tested for presence/absence of C. jejuni and C. coli by PCR. DNA was extracted from each of the faecal samples using the DNeasy PowerLyzer PowerSoil kit (#12855-50, Qiagen, UK). Primers were used to amplify portions of the mapA gene (C. jejuni) and cueE gene (C. coli) (Best et al., 2003), using the following thermocycling conditions; denaturation at 94°C for 2 min, primer annealing at 58°C for 1 min, and extension at 72°C for 1 min, for 35 cycles. PCR products were run out on an electrophoresis gel stained with SYBRTM Safe DNA gel stain (Thermo Fisher, UK) and compared with a 1KB DNA electrophoresis ladder.


All samples were processed in the laboratory within 24 hours of collection. For each sample, 200g faeces was mixed with 50ml of PBS and massaged for 1 minute. For samples 1-15, 2.5ml of the resulting suspension was added to 20ml of Bolton Enrichment Broth which was incubated at 41°C +/- 1°C for 48 hours. Each broth was then subcultured onto mCCDA and incubated microaerobically at 41°C +/- 1°C for up to 5 days. For samples 16-43, 2.5ml suspension from each was added to (i) Brain Heart Infusion Broth and (ii) Bolton Enrichment Broth which were then incubated at 41°C +/- 1°C for 48 hours. Each broth was then subcultured onto Skirrows Agar and mCCDA agar and incubated microaerobically at 41°C +/- 1°C for up to 5 days. In addition to the enrichment broth cultures,10µl of each of the sample suspensions were inoculated directly onto Skirrows Agar and mCCDA and incubated microaerobically at 41°C +/- 1°C for up to 5 days. The additional methods were introduced in case Campylobacter from horses had unusual or specific growth requirements or were present in low numbers, whilst maintaining consistency with other AgriFood, animal and environmental isolates by using mCCDA. We also included the method used by the veterinary clinical laboratory. Ten samples were tested by both sets of methods, giving the same results.
The isolates from the veterinary clinic were originally isolated using Bolton Broth and Skirrows Agar. They had been frozen down in Brain Heart Infusion Broth and 20% glycerol, and were recovered using Columbia Blood Agar, grown microaerobically at 41°C +/- 1°C for 48 hours.
The isolates from the veterinary clinic were included in the sequencing pipeline for the other AgriFood isolates, as previously described.
6.2.3.3. Results and discussion
Despite using the method that had successfully isolated Campylobacter from health screens of hospitalised horses, Campylobacter were not isolated using culture from any of the samples from healthy horses, nor were they detected by PCR. This included samples from horses grazing on recently flooded land, or horses under additional stress of staying away from home at a training camp and mixing with other horses. We used mCCDA upon which all the other AgriFood Campylobacter were isolated, as well as non-selective and selective enrichment broths designed to isolate Campylobacter from sub-optimal conditions, Skirrows agar from which Campylobacter were isolated from sick horses and a prolonged incubation time which can help to recover slow growing Campylobacter.
The sick horses from which Campylobacter was isolated had complex health conditions and it is likely Campylobacter was secondary to the main cause. The STs isolated were (i) ST-2412 (ST-45CC), recorded on PubMLST on only one other occasion from a chicken in the UK pre-2010, and (ii) ST-855 (ST-828CC, C. coli), also recorded predominantly from chicken and human disease, and from three different continents spanning the years 1994-2024. Both ST-45 and ST-828 clonal complexes can be found in multiple host sources indicating they may be adaptable to colonising horses even if they are not a primary host. They also had potential to cause human disease, the C. coli ST-855 having been previously recorded to cause human infection.
Results from this study indicate that healthy horses in the UK are an unlikely source of Campylobacter for their owners/carers or for waterborne outbreaks where other livestock/agricultural run-off or wild birds likely represent a greater risk. Campylobacter in sick horses could perhaps reflect infection from diffuse environmental contamination and/or gut dysbiosis associated with sickness that could lead Campylobacter to have higher prevalence than usual. Since isolates were required for WGS in this study and in the interests of cost and time, it was decided that efforts would be best directed to other more likely sources of infection.
6.4. Source attribution
Understanding the major reservoirs and transmission routes of Campylobacter is essential for developing effective control interventions. Over the past two decades, attribution studies have consistently identified poultry, particularly chicken, as the dominant reservoir of C. jejuni infection (Cody et al., 2019; Harrison et al., 2021; Pascoe et al., 2024; Thystrup et al., 2025). Molecular epidemiology has enabled increasingly precise source tracking, particularly with the adoption of multilocus sequence typing (MLST) and, more recently, whole-genome sequencing (WGS). These methods have been instrumental in revealing host-associated genetic signatures and informing attribution models.
Recent work has emphasised the need to treat C. jejuni and C. coli separately, given their distinct population structures, recombination patterns, and ecological associations (Jehanne et al., 2020).
Here, we apply a genome-informed machine learning (ML) model ‘aiSource’ (Arning et al., 2021) to attribute 10,477 C. jejuni and 1,165 C. coli isolates from Oxfordshire human disease (2003-2024) to likely non-human reservoirs, using publicly available genomes from PubMLST. Our approach integrates core genome MLST (cgMLST v2; 1,142 loci) and an ensemble of five machine learning classifiers (Random Forest, linear regression, xgBoost, LGMboost & catboost models) trained on high-quality reference genomes from known animal and environmental sources (n=27,431 C. jejuni and n=21,161 C. coli).
6.4.1. Training Dataset Compilation
All available non-human C. jejuni genomes were retrieved from the PubMLST database and filtered to include only assemblies with <300 contigs and valid source labels. The resulting dataset included isolates from multiple source categories, which were collapsed into unambiguous groups:
-
Chicken
-
Ruminants
-
Wild birds
-
Pigs
-
Other animals
Core genome MLST (cgMLSTv2) allele profiles were extracted using the 1,142 locus scheme. Isolates with >20% missing data were excluded. This dataset was used to train the source attribution model.
6.4.2. Model training and evaluation
Training data were partitioned (75:25) to train and test the accuracy of each ML classifier compared to the smaller partition of blinded isolates, which were mock predicted to a potential source. For both species the CatBoost classifier performed best. For C. jejuni, a self-test accuracy was 86.92%, before 100 bootstraps were used to assess model accuracy, giving a mean of 91.00% accuracy. For C. coli isolates, the self-test accuracy was 87.19%, and after 100 bootstraps, this yielded a model accuracy with a mean of 90.60%. This is consistent with results obtained using aiSource with isolates from US public health, where the CatBoost ML classifier also performed best (Pascoe et al., 2024).
6.4.3. Source attribution pipeline
To perform high-resolution source attribution, we developed and applied a custom machine learning pipeline written in Python, based on the published aiSource method (Arning et al., 2021). The pipeline integrates core genome MLST data with supervised classification models to infer the most likely source of Campylobacter isolates. The design emphasizes accuracy, reproducibility, and extensibility for public health and research settings.
The pipeline consists of two key components:
-
The main orchestration script that handles data ingestion, filtering, imputation, model training, prediction, and visualization. It supports flexible command-line execution and is designed to operate on large datasets with cgMLST allele profiles and associated metadata.
-
A companion module containing utility functions for classifier selection, under/oversampling (e.g., SMOTE), imputation, model evaluation, and figure generation. It also supports optional modules for SHAP explainability and Plotly-based interactive visualizations.
Pipeline Features and Capabilities:
-
Model selection and ensembling: supports multiple classifiers including XGBoost (default), Random Forest, LightGBM, CatBoost, and Logistic Regression. It can automatically select the best-performing model via cross-validation or use ensemble strategies (e.g., soft voting or stacking).
-
Data preprocessing and imputation: The pipeline filters input allele matrices based on a user-defined missingness threshold (default: 20%) and applies either simple or phylogeny-aware imputation to fill in missing data.
-
Balanced training with resampling: To mitigate class imbalance, the pipeline supports under- and oversampling techniques, including SMOTE if available. This improves model generalisability across underrepresented source categories.
-
Bootstrapping and uncertainty propagation: The pipeline includes a bootstrapping module that reruns predictions across multiple resampled models to estimate the stability of attribution outcomes and generate confidence intervals for each prediction.
This comprehensive framework underpins all attribution analyses presented in this report and enables flexible, reproducible genomic epidemiology of C. jejuni and C. coli. It can be readily adapted for other pathogens or data types where source attribution or host prediction is required.
6.4.4. Attribution of Oxfordshire human disease Isolates
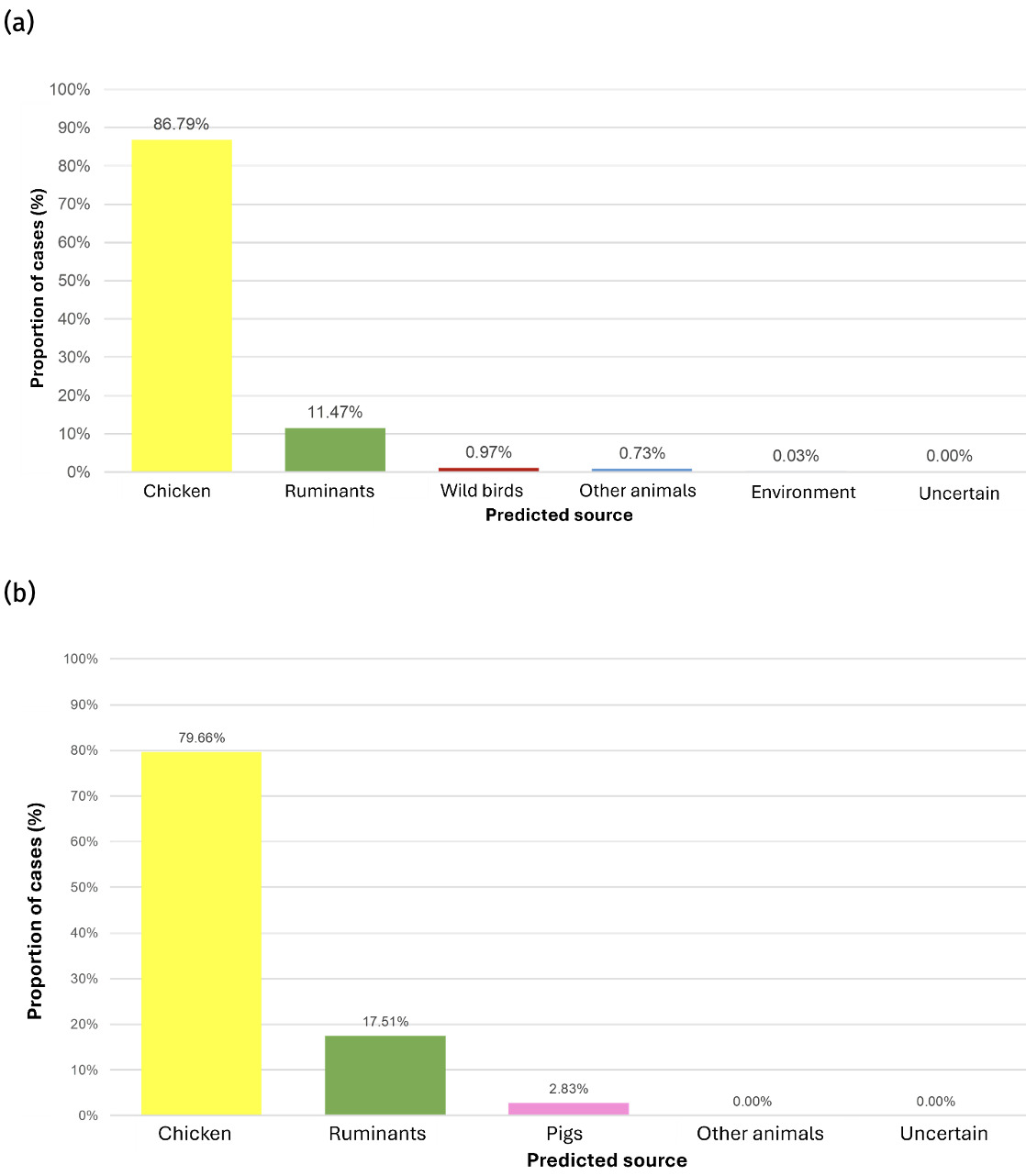
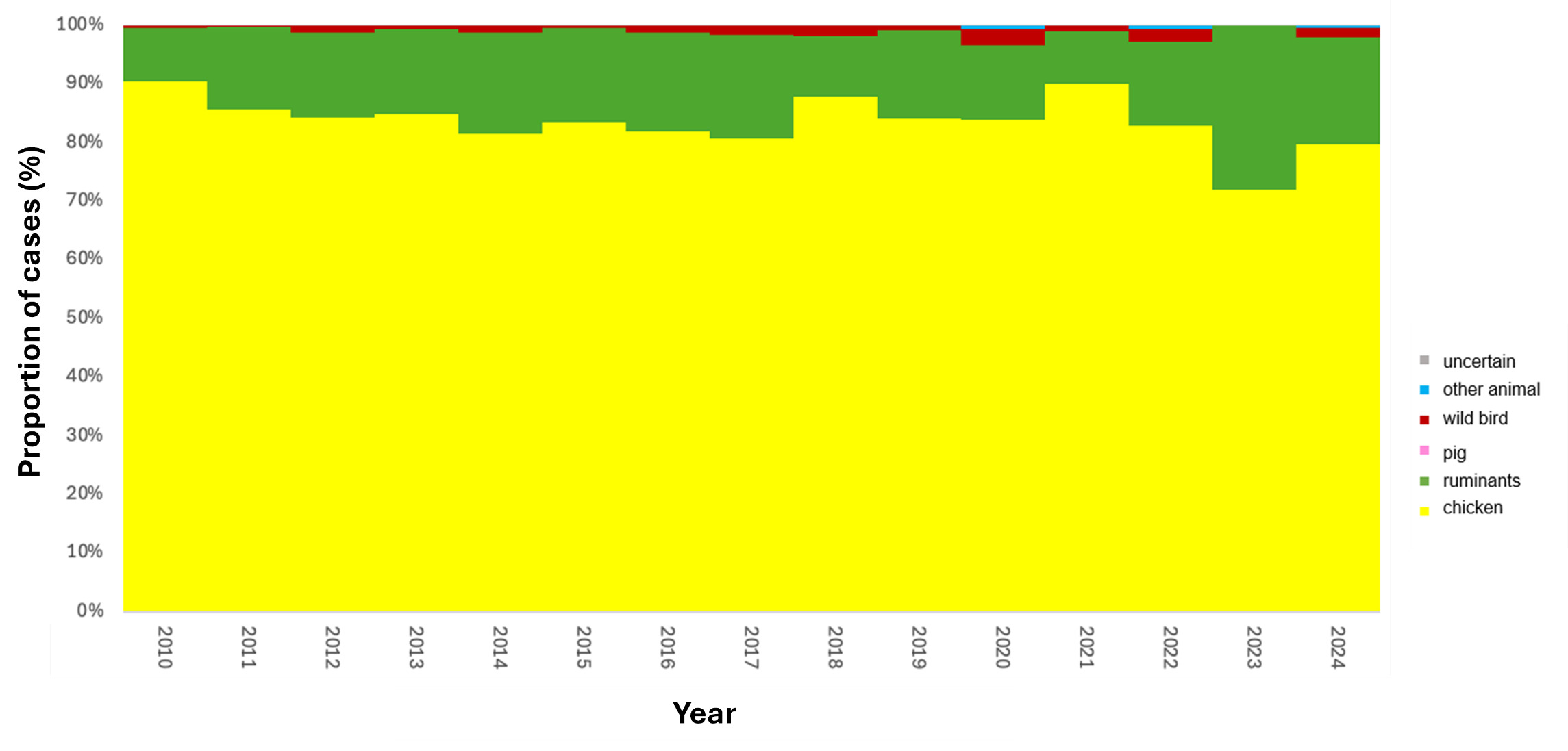
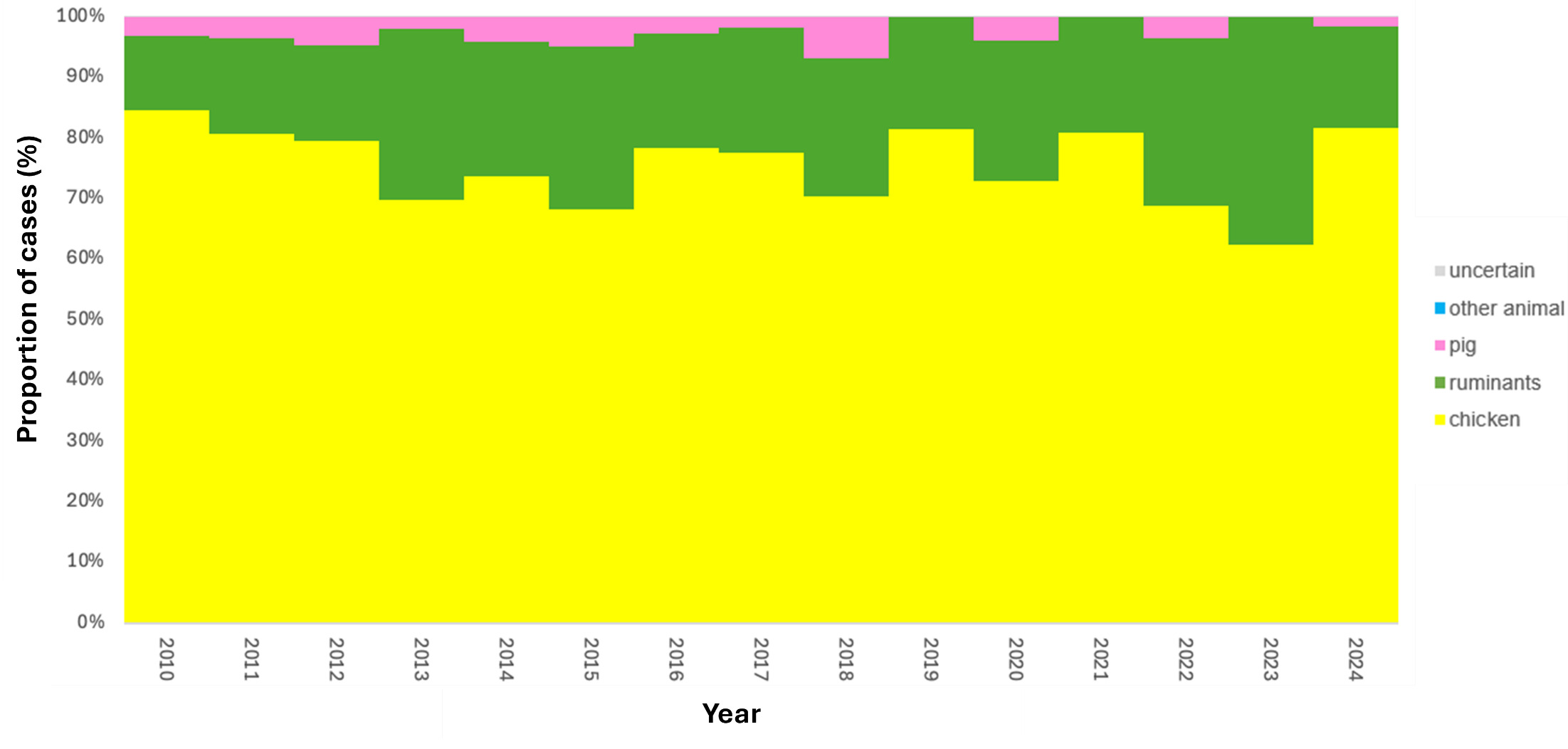
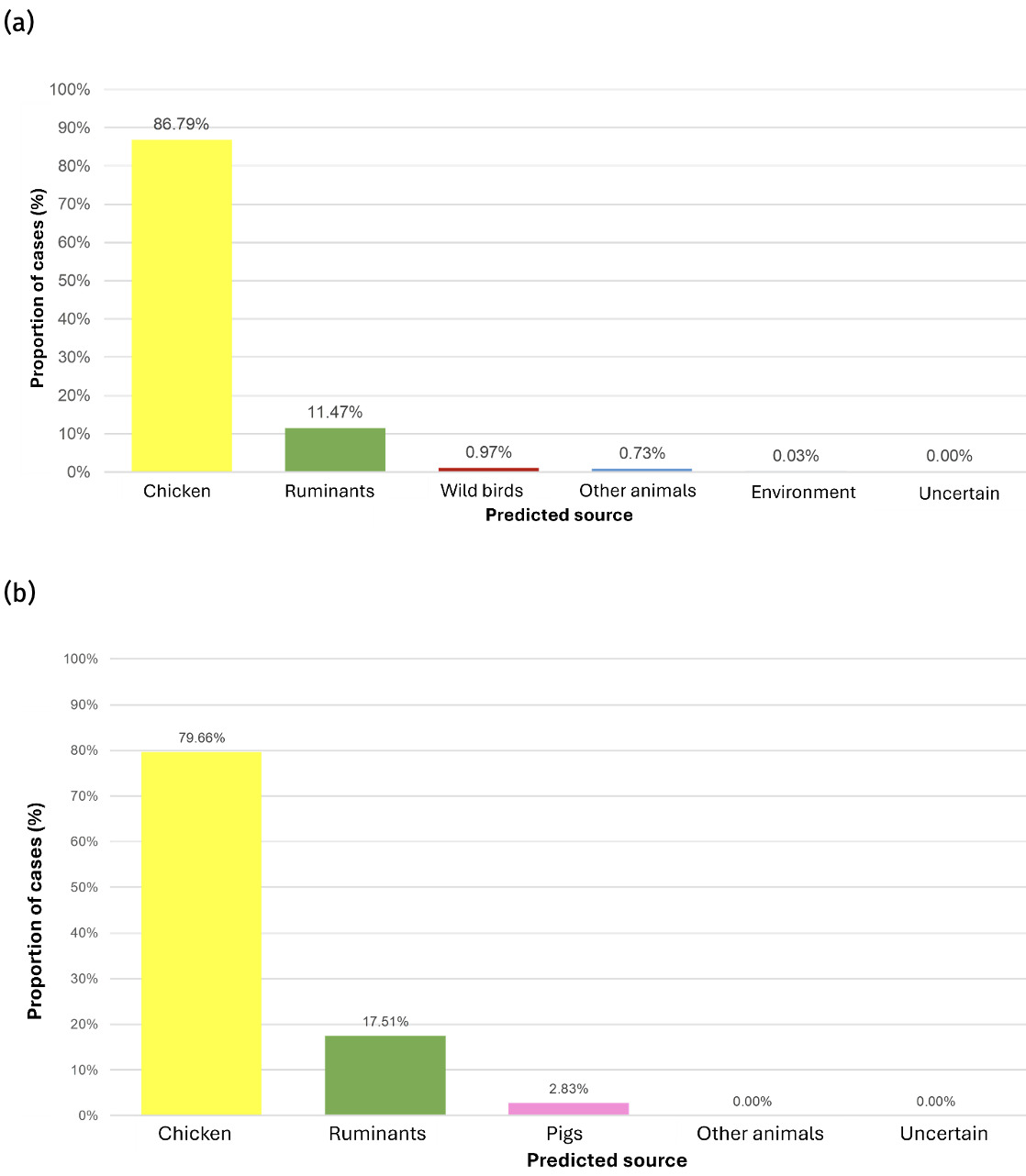
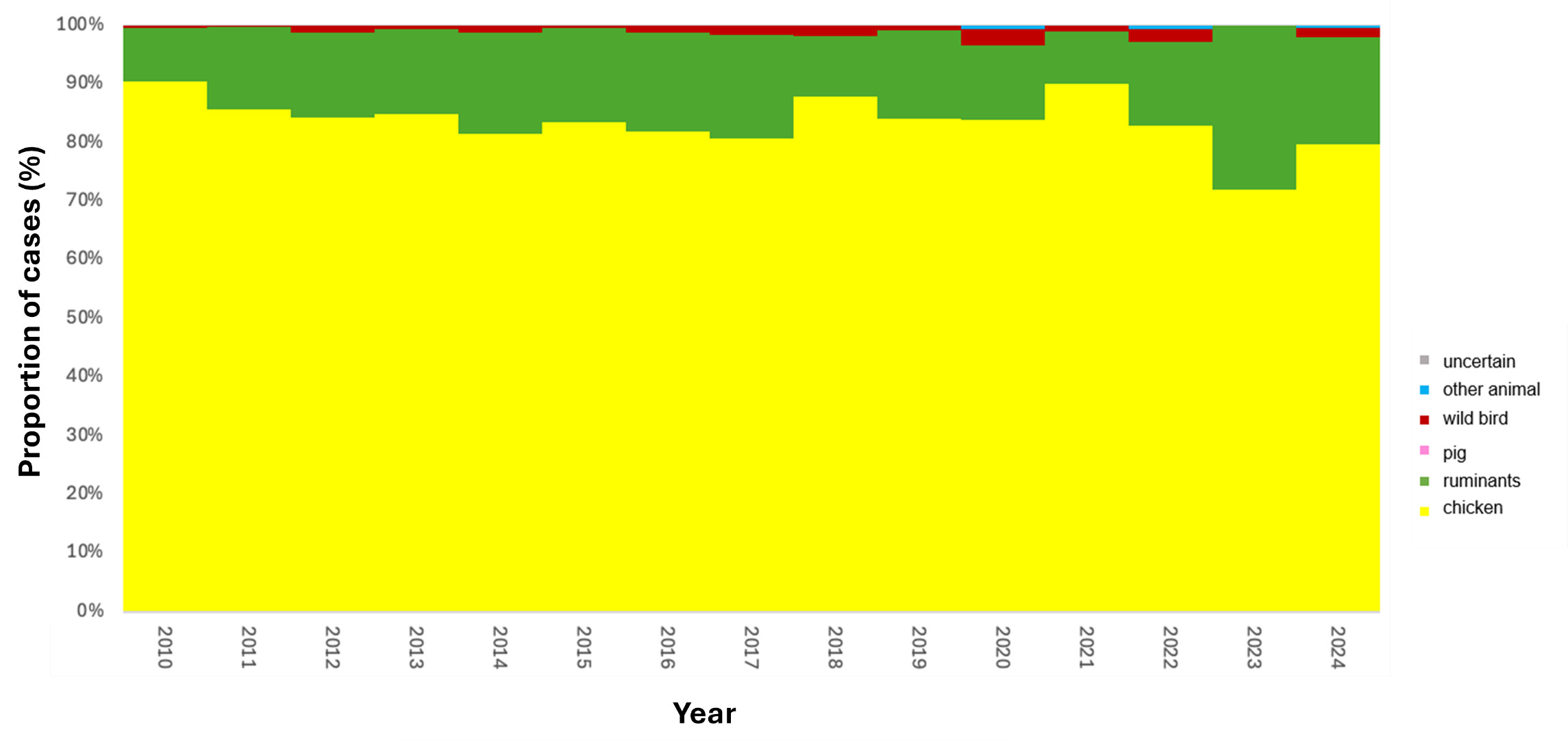
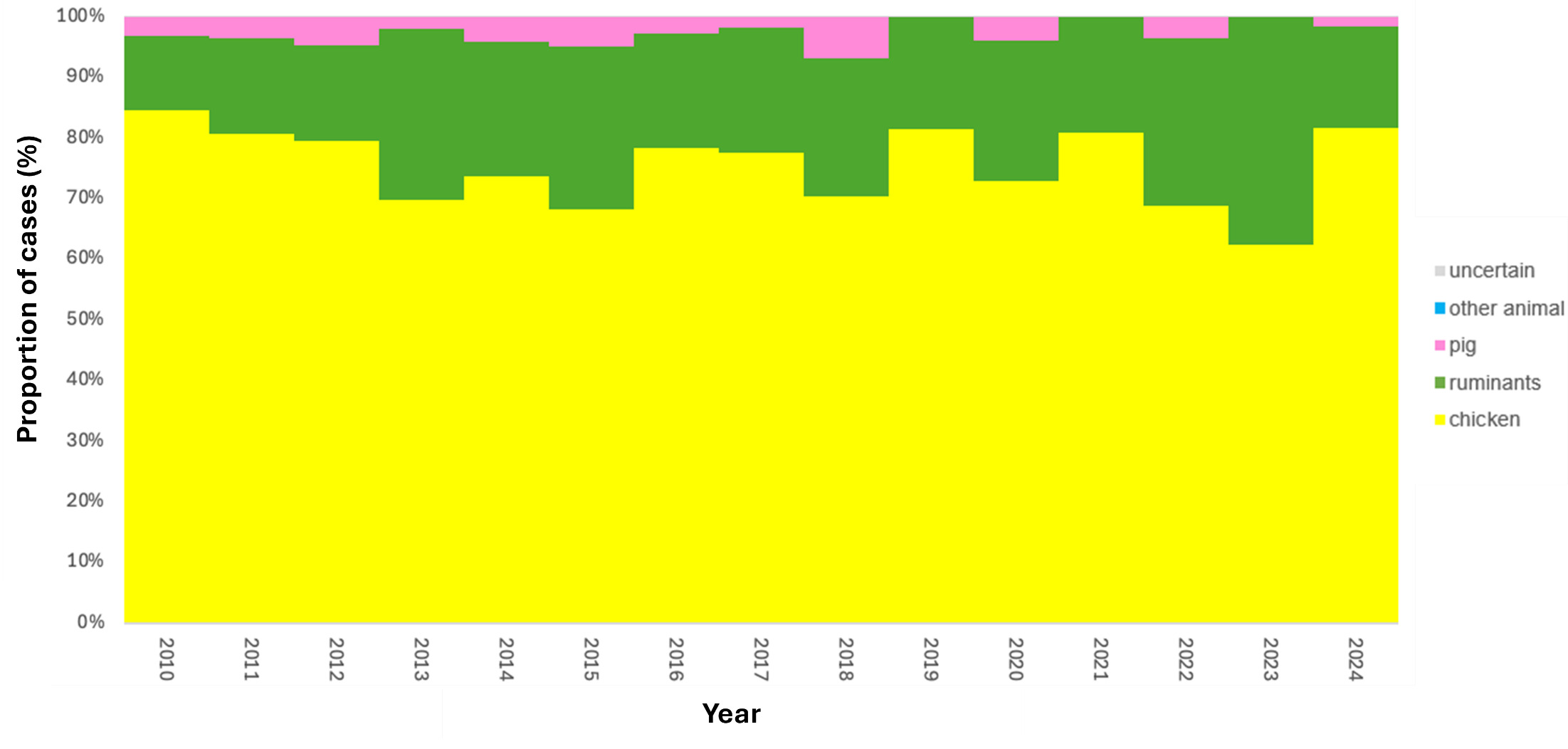
The trained model was applied to 11,642 genomes from Oxfordshire human disease (2003-2024) (n=10,477 C. jejuni and n= 1,165 C. coli). Each isolate was assigned a predicted source with associated class probabilities. Results are summarised for the whole collection of Oxfordshire human disease isolates (Figure 10) and by year (Figure 11). The majority of human disease isolates were predicted to come from chicken for both C. jejuni (86.7%) and C. coli (79.7%), followed by ruminant sources (C. jejuni 11.5%, C. coli 17.5%). Despite a dip in 2023, there is no notable, sustained change in source attribution longitudinally.
6.4.5. Discussion
Our findings reaffirm the central role of chicken as the predominant source of C. jejuni and C. coli infection in the UK, consistent with prior studies using MLST and STRUCTURE-based approaches (Cody et al., 2019; Oxford, 2019; Sheppard et al., 2009). The contribution from ruminants is also considerable, shown to be particularly important in rural or agricultural regions in previous studies (Pascoe et al., 2024; Strachan et al., 2013). Wild birds and environmental sources appear to play a minor role in human Campylobacter disease, although these may be underrepresented in the training data.
The use of cgMLST and machine learning allowed for high-resolution source attribution, improving on the discriminatory power of traditional MLST. Our decision to analyse C. jejuni independently of C. coli reflects recent evidence that mixed models can obscure true host associations due to differing evolutionary dynamics (Jehanne et al., 2020).
Limitations include possible sampling biases in the reference data and uneven coverage across source categories. Nonetheless, the model’s high performance and consistent trends support its utility for routine surveillance and intervention targeting.
This genomic attribution analysis highlights the persistent dominance of chicken in C. jejuni and C. coli transmission to humans in the UK. Routine incorporation of WGS and AI-based attribution frameworks into public health surveillance could support more targeted control measures across the food production chain.
6.5. Genomic prediction of antimicrobial resistance in Campylobacter isolates
6.5.1. Fluoroquinolone and tetracycline resistance; human disease isolates.
Isolates with at least one genetic determinant predicting resistance to either fluoroquinolones and/or tetracycline accounted for 1331/2324, 57.3% of the human disease C. jejuni isolates from Oxfordshire (2019-2024) and Swansea (2012-2013), shown in Table 11. Considering the antimicrobials individually, 1174/2324, 50.5% of the human disease isolates were predicted to be resistant to fluoroquinolones and 931/2324, 40.1% of the isolates were predicted to be resistant to tetracycline. Thr-86-Ile was the most common determinant predictive for fluoroquinolone resistance (n=1170/2324, 50.3%), of which 771/1170, 65.9% also carried the tet(O) gene predicting resistance to tetracycline. Other single point mutations, Thr-86-Val and Thr-86-Ala were rare, accounting for 1-2 isolates (0.04-0.09%) each respectively. A double mutation Thr-86-Ile and Pro-104-Ser was identified in 68/2324 (2.93%) isolates, of which one had a tet(O) gene present also. A second isolate with a double gyrA mutation of Thr-86-Ile and Asp-90-Asn (D-90-N), also resistant to tetracycline, was also detected. A total of 157/2324, 6.76% of isolates carried tet(O) without resistance to fluoroquinolones.
The most common predictive determinant for C. coli fluoroquinolone resistance was also Thr-86-Ile (n=94/304, 30.9%), of which 56.4% (n=53/94) also carried the tet(O) gene (Table 12). Only one other single point mutation (Thr-86-Val) was found in one isolate, and no double point mutations conferring fluoroquinolone resistance were detected amongst the human disease C. coli isolates.
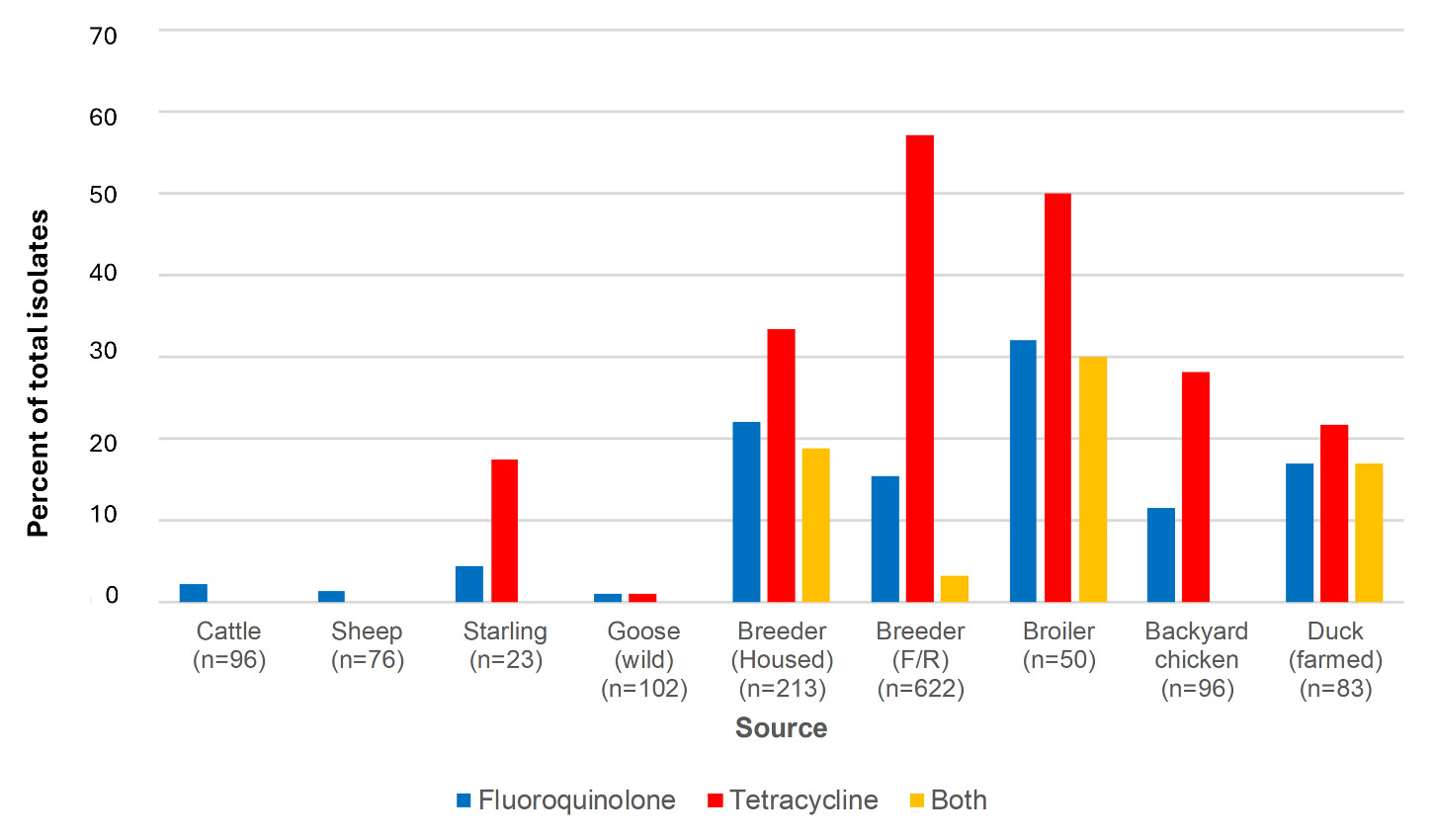
6.5.2. Fluoroquinolone and tetracycline resistance; AgriFood and environmental isolates.
As with the human isolates, the Thr-86-Ile point mutation conferring fluoroquinolone resistance was most common amongst both C. jejuni (n=134/862, 15.5%) (Table 13) and C. coli (n=53/520, 10.2%) (Table 14) isolated from the AgriFood and environmental sources. One other single point mutation Pro-104-Ser was detected amongst the C. coli isolates (n=1/520, 0.19%) and one double point mutation Thr-86-Ile and Pro-104-Ser (n=1/862, 0.12%) was detected amongst the C. jejuni isolates. The tetO gene predicting resistance to tetracycline was detected amongst 261/862 (30.3%) C. jejuni and 217/520 (41.7%). C. coli isolates from animal and environmental origin (Tables 13 and 14). Resistance to both antibiotic classes was predicted for 45/862, 5.22% of C. jejuni and 50/520 (9.62%) of C. coli isolates.
6.5.3. Macrolide resistance; all isolates from human disease (Oxfordshire 2019-2024 and Wales 2012-2013) and AgriFood and environment isolates.
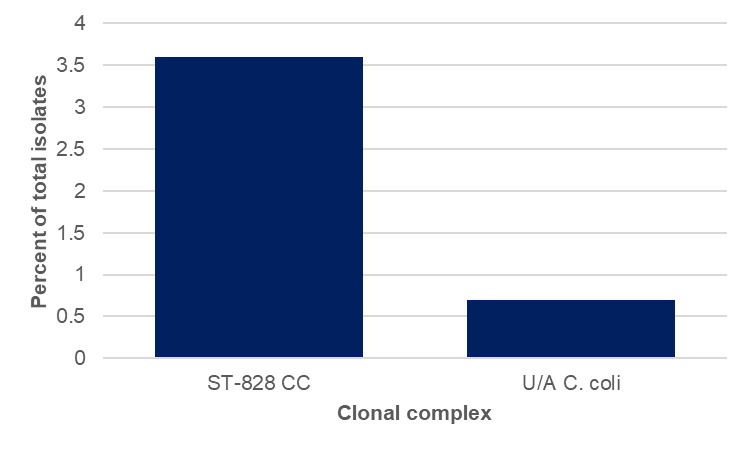
Amongst the human disease isolates, macrolide resistance was predicted for 13 C. coli isolates (13/300, 4.3%), 11 of which grouped into the ST-828CC with two isolates unassigned (Figures 12 and 13). The A2075G mutation was identified amongst all of the resistant isolates. Nearly every isolate was a different ST, with the exception of ST-872 and ST-1055 which were both isolated on 2 occasions. The isolates with a 23s rRNA A2075G mutation were detected in 2019 (n=4), 2020 (n=1), 2021 (n=1), 2022 (n=2), 2023 (n=2) and 2024 (n=3).
No macrolide resistance was detected amongst the Oxfordshire human disease C. jejuni isolates, or either Campylobacter species from the AgriFood and environment isolates.
The emerging ermB gene (Bolinger & Kathariou, 2017) conferring high level macrolide resistance was not detected amongst any isolates in this study.

6.5.4. Aminoglycoside resistance; all isolates from human disease (Oxfordshire 2019-2024 and Wales 2012-2013) and AgriFood and environment isolates.
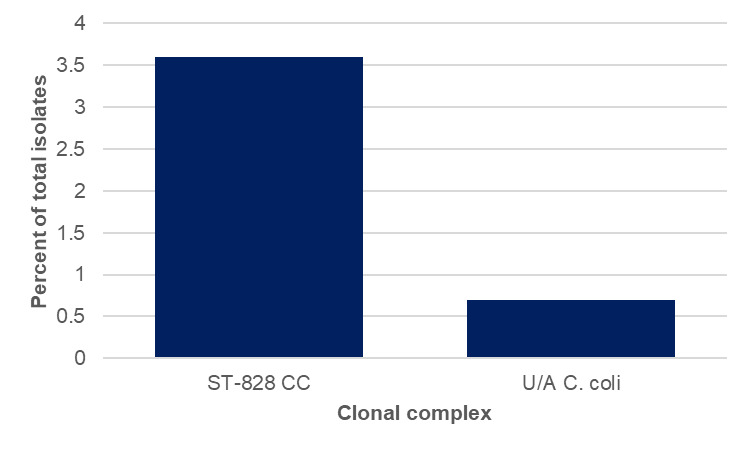
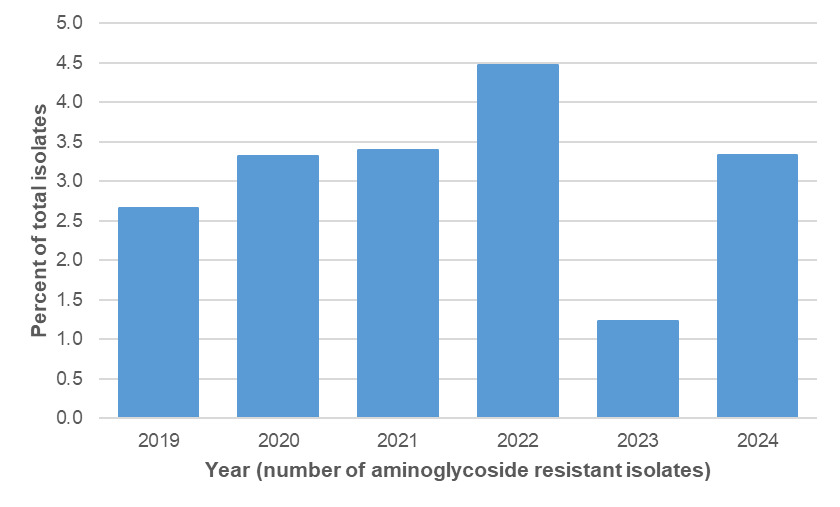
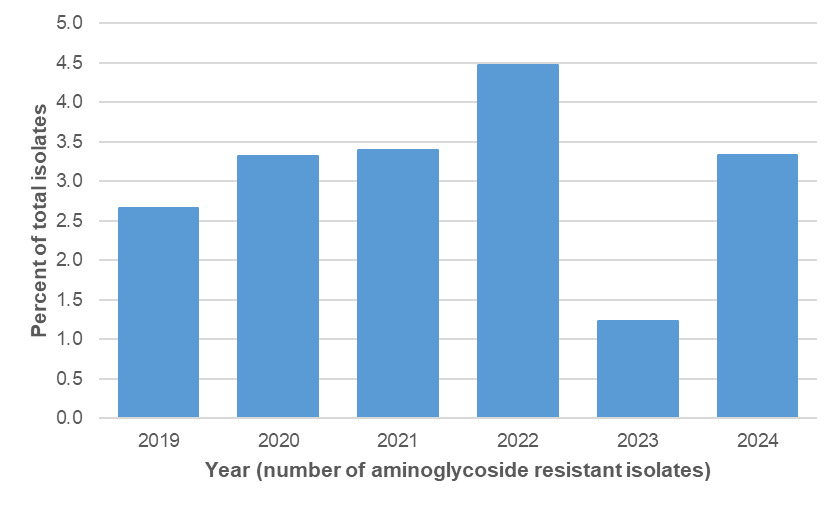
Amongst the Oxfordshire human disease isolates with genetic determinants for aminoglycoside resistance were detected amongst 45/2363 (1.9%) C. jejuni and 36/300 (12.0%) C. coli isolates, using PubMLST (Table 15). The distribution by year is shown in Tables 16 and 17 and Figure 14 below. Aminoglycoside resistance determinants were identified amongst ten C. jejuni clonal complexes; ST-353CC (n=17), ST-42CC (n=7), ST-206CC (n=7), (ST-21CC (n=3), ST-45CC (2), ST-48CC (n=2), ST-257CC (n=2), ST-607CC (n=2), ST-354 (n=1), ST-464CC (n=1) and two unassigned C. jejuni isolates (Figure 15). For C. coli, aminoglycoside resistance determinants were identified amongst 73 ST-828CC isolates, one ST-1150CC isolate and 24 unassigned C. coli isolates.
Aminoglycoside resistance was detected amongst only one C. jejuni AgriFood isolate from cattle. The isolate was ST-42, grouping into the ST-42 clonal complex, with the rpsL K43R mutation associated with aminoglycoside resistance. It was otherwise sensitive to the other antibiotics tested in this study. An Oxfordshire human disease isolate with the same ST and resistance profile was isolated in 2021.
Aminoglycoside resistance was detected amongst 59 C. coli isolates from housed and free-range broiler breeder chickens, 2 sheep and 1 starling isolate. All had the aadE-Cc determinant, with the exception of one free-range broiler breeder chicken isolate (ST-7159, ST-828CC) which had the ant(6)-1a determinant.

6.5.5. Multidrug resistance; all isolates from human disease (Oxfordshire 2019-2024 and Wales 2012-2013) and AgriFood and environment isolates.
Amongst the human disease C. jejuni isolates, 13 were resistant to three or more antimicrobial categories tested in this study, identified in the years 2019 (n=4), 2020 (n=4), 2021 (n=2) and 2022 (n=4). They were most commonly from ST-353CC (n=7), followed by ST-48CC (n=2), and one isolate each from ST-21CC, ST-206CC, ST-354CC, ST-464CC and one unassigned isolate. All were resistant to fluoroquinolones, tetracycline and macrolides. Amongst the human disease C. coli isolates, nine were resistant to fluoroquinolones, tetracycline and aminoglycosides, one was resistant to fluoroquinolones, macrolides and aminoglycosides, and one was resistant to fluoroquinolones, tetracycline, macrolides and aminoglycosides (ST-1109, unassigned to a clonal complex).
Amongst the AgriFood and environmental C. jejuni isolates, none were resistant to three or more antimicrobial categories tested in this project. Twenty C. coli isolates, all ST-828 (ST-828CC) from the free-range broiler breeder flock, were resistant to fluoroquinolones, tetracycline and aminoglycosides. No AgriFood and environmental isolates were resistant to all four antimicrobials tested in this study.
6.5.6. Resistance Enhancing cme (RE-cmeABC) multidrug efflux pump; all isolates.
Mutation in the CmeR repressor protein binding site for the transmembrane cmeABC efflux pump has been associated with over expression of the pump and increased resistance to ciprofloxacin, erythromycin and multidrug resistance (Schiaffino et al., 2024; Yao et al., 2021). The CmeR binding site is an inverted repeat sequence of around 15-22 nucleotide base pairs in the promoter region upstream of the cmeA gene. The CmeR binding site sequence, including flanking sequence and the additional CosR repressor protein binding site sequence is named proCAMP0332 and is searchable on PubMLST. In addition, variation in the cmeB gene has been associated with a 4-fold increase in ciprofloxacin (fluoroquinolone) resistance and 9-fold increase in erythromycin (macrolide) resistance.
A total of 287/4010 (7.16%) isolates in this study were found to have mutations in the CmeR binding site. They were most commonly identified amongst human disease isolates 205/287, 71.4%, with the remainder isolated from chicken (69/287, 24.0%), geese (12/287, 4.2%) and 1 starling (1/287, 0.3%) (Figure 16, Table 18 and Table 29, appendix A). Amongst Oxfordshire human disease isolates, isolates predicted to have cmeR determinants associated with efflux pump overexpression accounted for 21-40 (6.4-9.7%) of isolates each year, from 2019-2024, and 202/2585, 7.8% overall.
The CmeR binding site mutations were identified amongst 19 clonal complexes, of which ST-21CC was the most common (215/287, 74.9%), followed by ST-353CC (13/287, 4.5%), ST-206CC (15/287, 5.2%) and ST-1034CC (11/287, 3.8%) (Figure 17). The remaining clonal complexes had fewer than four isolates with CmeR binding site mutations each. A total of 15 different mutations in the CmeR binding site were identified, 6 of which have been previously identified amongst isolates with increased AMR from Peru and China, and 9 newly identified in this study (Schiaffino et al., 2024; Yao et al., 2021). Most, but not all, isolates also contained the Thr-86Ile mutation conferring resistance to fluoroquinolones, but further work is needed to confirm the level of the AMR phenotype of the UK isolates. Just two human disease isolates (from 2019 and 2023) with CmeR binding site mutations had additional genetic resistance determinants to fluoroquinolones, tetracycline and macrolides.
The cmeB protein is an inner membrane transporter, and structural modelling suggests that variants with approximately 81% amino acid sequence identity to cmeB in C. jejuni 11168 may result in enhanced function (Yao et al., 2016). Variant cmeB genes were found in 160/4010 (4.0%) of isolates in the study, most commonly amongst human (n=51) and chicken (n=106) isolates, with two from cattle, and one from a farm environment source. The cmeB variant genes were not always found with cmeR binding site or gyrA mutations, and the combination of all three were not commonly found together in the same isolate (Table 18).
Taken together, presence of either or both of the cmeR or cmeB resistance elements were found amongst 439/4010 (10.9%) of isolates across the study. They were most commonly identified in C. jejuni (n=435) isolates compared to C. coli (n=4) isolates, and from human disease (n=251) and chicken (n=172) compared to geese (n=12), cattle (n=3), and the farm environment (n=1). Amongst the Oxfordshire human disease isolates they accounted for 34-52 (8.6-10.2%) C. jejuni isolates per year, and 1 C. coli isolate each in years 2020, 2021, 2023 and 2024. Further work is needed to confirm how the RE-cmeABC genotype predicts phenotype for UK isolates.


6.6. Antimicrobial resistance phenotyping
Although phenotype is generally found to correlate well with predictive AMR genotype for Campylobacter, we tested this more extensively amongst clinical isolates across the diversity of lineages (clonal complexes) and AMR resistance determinants.
A subset of 130 isolates (112 C. jejuni and 18 C. coli) from the extended Oxfordshire human disease isolate collection 2010-2024, were selected for AMR phenotyping. They were chosen to test resistance across the different lineages (clonal complexes) and resistance determinants, but were otherwise chosen at random from isolates with high quality whole genome sequencing data. The minimum inhibitory concentration (MIC) was determined using E-tests following the 2025 EUCAST guidelines (https://eucast.org), with results interpreted as ECOFFs. The isolates were grown on Mueller Hinton agar with 5% horse blood and NAD, incubated at 41°C +/- 1°C and the results read at 24 and 48 hours.
A total of 112 isolates were tested phenotypically for ciprofloxacin (fluoroquinolone) resistance (Table 19). All except the isolates from six different clonal complexes with the wild type gyrA genotype recorded MICs (< 0.0125 mg/L) and were below the ECOFF (Epidemiological cut-off for antimicrobial resistance phenotyping) for resistance (0.5mg/L) but above the cut-off for sensitivity (≤0.001mg/L). Following EUCAST guidelines, the isolates were classed as susceptible, increased exposure to fluoroquinolones, meaning there would be high likelihood of therapeutic success by adjusting the dosing regime. One of 74 isolates (1.4%) with the gyrA Thr-86-Ile mutation predicted to be resistant to ciprofloxacin was sensitive upon testing (MIC 0.064). This isolate grouped into the ST-45 clonal complex and was isolated in 2012. The rest of the isolates tested with the Thr-86-Ile were resistant (MIC > 3mg/L) as predicted, with most recording MICs of 32mg/L, the highest level tested. The rarer D90N and P104S gyrA mutations were also phenotypically resistant, with MICs of 8 and 32 mg/L respectively. Overall, fluoroquinolone resistance determinants showed excellent correlation with resistance phenotype across the different lineages (p <0.05).
A total of 62 isolates were chosen to represent different clonal complexes, including biological replicates where possible, and tested phenotypically for tetracycline resistance (Table 20). All of the isolates from nine clonal complexes predicted to be resistant due to the presence of the tet(O) gene were resistant, as expected, with MICs ranging from 4mg/L to 256mg/L. Of these, 20/33 (60.1%), from eight different clonal complexes had the highest resistance level of 256mg/L. One isolate (1/62, 1.6%) that was predicted to be sensitive to Tetracycline as the tet(O) gene was not detected from the whole genome sequencing data was resistant with an MIC of 64mg/L. This isolate was grouped into the ST-257 complex and was isolated in 2015. Overall, phenotypic tetracycline resistance showed excellent correlation with genotypic prediction (p <0.05).
A total of 19 isolates were tested phenotypically for erythromycin (macrolide) resistance (Table 21) with nine isolates predicted to be resistant due to the 23S rRNA A2075G mutation and ten isolates with wild type determinants predicted to be sensitive. The resistant isolates were from five clonal complexes and unassigned C. jejuni and C. coli isolates, and they all recorded high resistance at 256 mg/L. This was despite the absence of the emerging ermB gene (Bolinger & Kathariou, 2017) that confers high level erythromycin resistance. The ten isolates predicted to be sensitive were indeed sensitive phenotypically, with MICs of <1 mg/L.
At the time of writing, (September 2025) there are no EUCAST guidelines regarding the MIC for aminoglycoside antibiotics for Campylobacter. Resistance determinants for the RE-cmeABC pump also require validation for UK isolates, which was unfortunately beyond the scope of this study.
6.7. Trends in Campylobacter antimicrobial resistance amongst human disease isolates over time
Following cross-validation with other platforms (see section 6.1.3.5 and our previous report [Oxford, 2019]), fluoroquinolone resistance was predicted genotypically by presence of mutations in gyrA, and tetracycline resistance predicted by presence of tet(O) using the PubMLST database. Note, fluoroquinolone resistance results are shown for isolates in which the full coding sequence of gyrA (CAMP0950) was identified. Tetracycline resistance (presence of the tetO gene) was determined amongst isolates with high quality sequencing data (cgMLST annotation = ‘good’ and 500 contigs or less). The macrolide and aminoglycoside resistance results are shown for all isolates, but were cross-checked against raw sequencing reads using ResFinderPlus to ensure that all copies of the 23s rRNA gene were accounted for. The genomic resistance determinants accurately predicted resistance phenotype amongst all except two isolates from the subset of 150 isolates tested in this study, and across a range of lineages (see section 7.6). The numbers of C. coli isolates are much lower than C. jejuni and may account for some of the stochasticity seen.
Data from Oxfordshire UK human disease isolates 2019-2024 in this study were compared with earlier human disease isolates from Oxfordshire (2003-2018), with resistance determinants identified in the same way using the PubMLST database. The data for 12,810 isolates can be viewed on PubMLST (https://pubmlst.org, project 1, Oxfordshire Human Surveillance); Oxfordshire Human Surveillance - Campylobacter jejuni/coli isolates. Note, a user account is needed to log in to view the isolates – account registration is free.
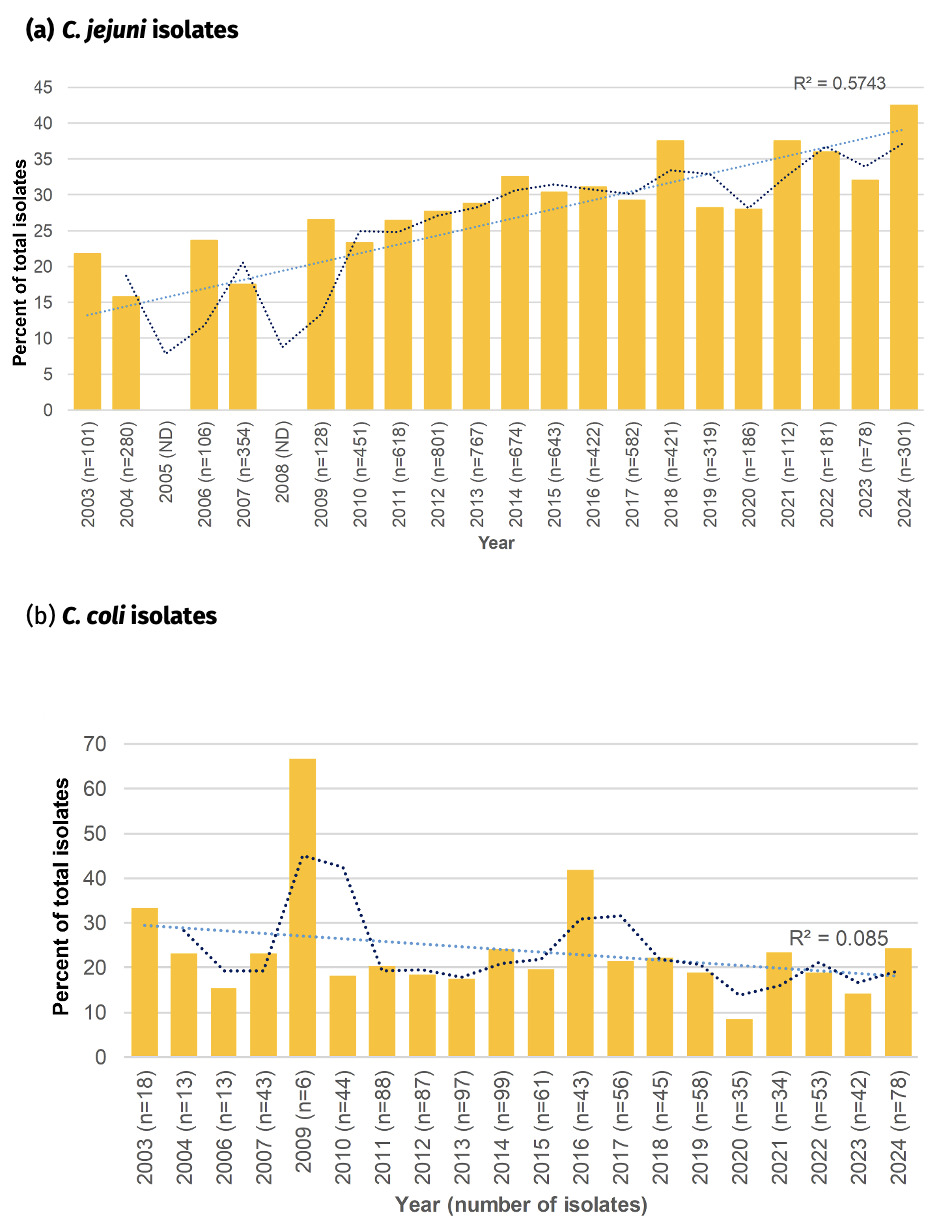
6.7.1. Fluoroquinolone resistance amongst Oxfordshire human disease isolates, 2003-2024
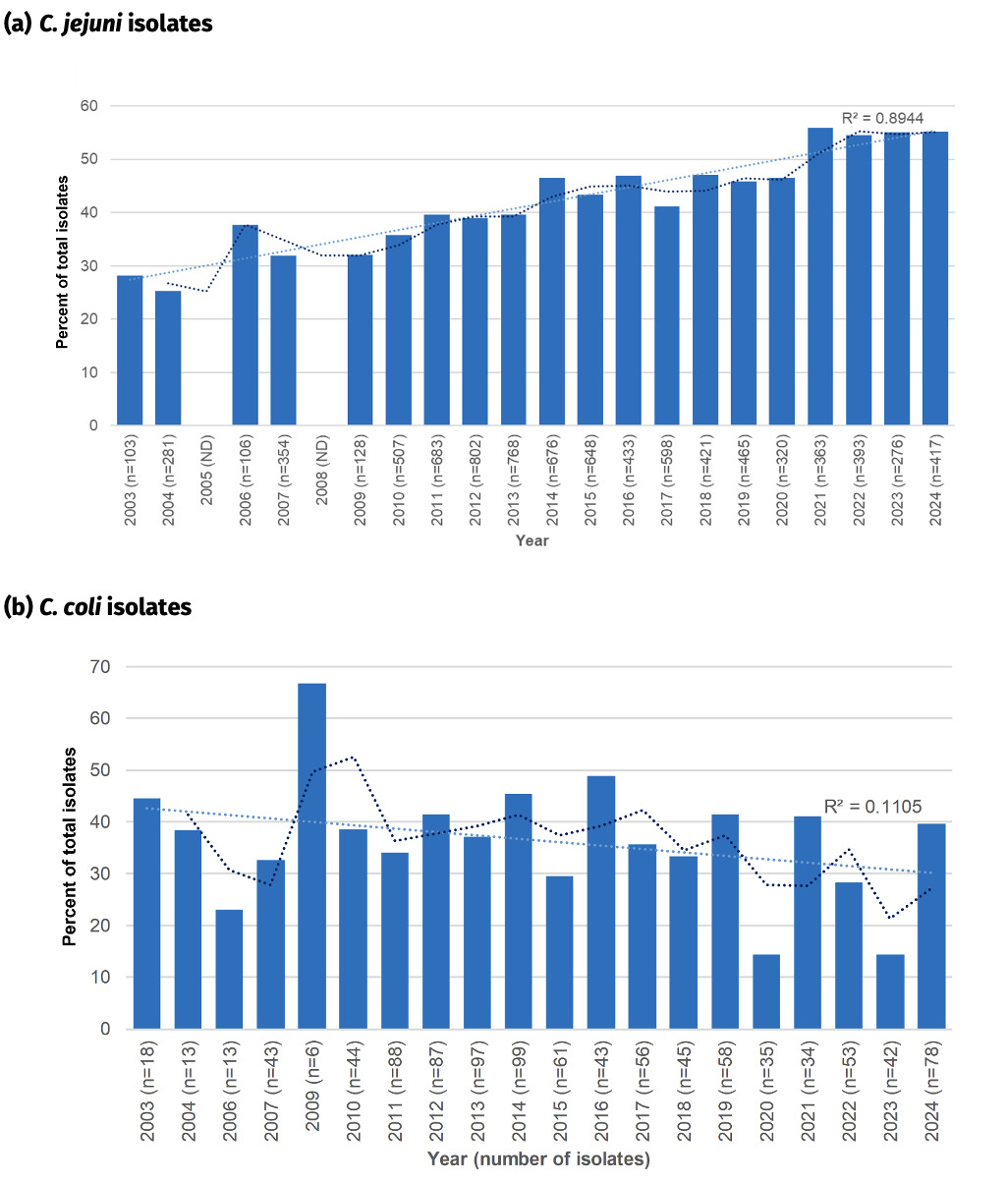
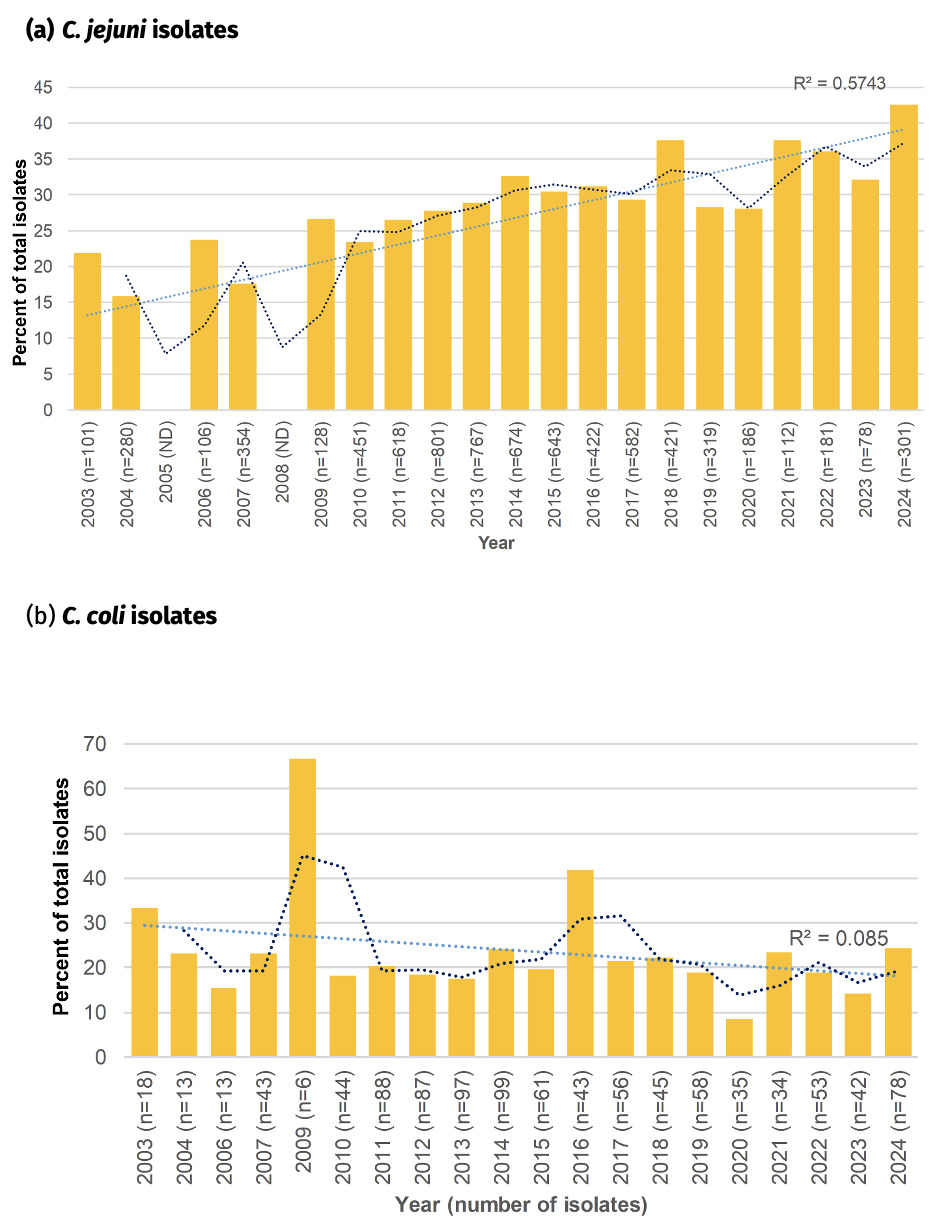
Fluoroquinolone resistance has almost doubled amongst Oxfordshire human disease C. jejuni isolates from 28.2% in 2003 to 55.2% in 2024 (Figure 18) and the linear trendline shows a good fit with time (R2 = 0.89). With a significant reduction in antimicrobial use on farms since 2012, the reason(s) for the continued increase in resistance is unknown and should be investigated further. The highest resistance of 55.9% was recorded in 2021. Resistance levels have been relatively stable over the past 3 years (54.5% to 55.2% in years 2022 to 2024), but similar plateaus and stepwise increases in rate of resistance acquisition, can be seen in the years 2011-2013 (~39% resistance) and 2014-2020 (~46% resistance), before an abrupt increase in 2021.
In comparison, fluoroquinolone resistance was higher amongst C. coli isolates in 2003 (44.5%), and has mostly remained between 30-45% resistance since then, with occasional exceptions. The highest level of resistance was recorded in 2009 (66.7%) and lowest in 2020 (15.6%). The linear trendline suggests a slight decrease in fluoroquinolone resistant C. coli over the time period, but is not well supported (R2 = 0.11).
_*c._jeju.png)
6.7.2. Tetracycline resistance amongst Oxfordshire human disease isolates, 2003-2024.
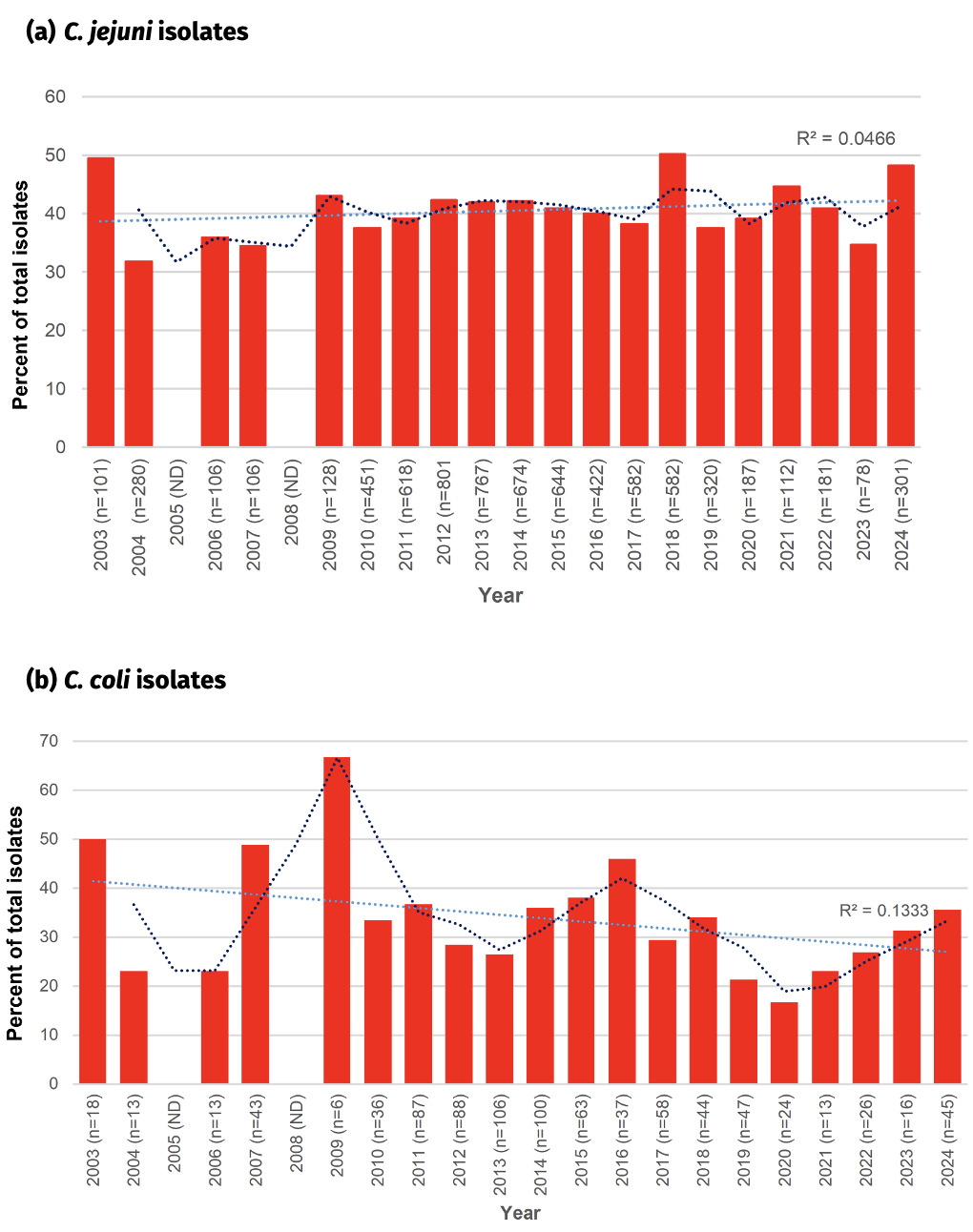
In contrast to fluoroquinolone resistance, tetracycline resistance has remained relatively stable amongst the Oxfordshire human disease C. jejuni isolates, with between 30-50% of isolates having the tetO gene encoding resistance (figure 19). Highest levels were recorded in 2003 (49.5%), 2018 (50.1%) and 2024 (48.2%), and lowest levels were recorded in 2004 (31.8%), 2007 (34.5%) and 2023 (34.6%). The linear trendline did not show a good fit with time (R2 = 0.05).
Tetracycline resistance was more varied amongst the C. coli isolates, perhaps in part due to the smaller number of isolates in some years. Resistance levels were most commonly between 20-40%, slightly lower than that seen amongst C. jejuni isolates. The linear trend line suggests a slight general downwards trend in resistance over time, but is not well supported (R2 = 0.13), and in fact over the past 5 years tetracycline resistance has more than doubled from 16.7% to 35.6%.
The overall 39.5% (534/1350) percentage of tetracycline resistance amongst Oxfordshire human disease isolates with highest sequencing quality (cgMLST annotation = ‘good’, </= 500 contigs) exactly matched that amongst all Oxfordshire human disease isolates, 1021/2585 (39.5%). Results are shown for isolates with highest sequencing quality.
_*c._jejuni*_.png)
6.7.3. Co-resistance to both fluoroquinolone and tetracycline resistance amongst Oxfordshire human disease isolates, 2003-2024
A high proportion of Campylobacter isolates were resistant to both fluoroquinolones and tetracycline, with trends resembling rising fluoroquinolone resistance in C. jejuni, whilst they were a little more stable amongst C. coli (Figure 20). The rise in co-resistance in C. jejuni was less clearly associated with a linear trend (R2 = 0.57) compared to fluoroquinolone resistance on its own. The highest prevalence was recorded in 2024 (42.5%), doubling in prevalence from in 2003 (21.8%). Co-resistance in C. coli was most commonly around 20% of isolates. An overall downwards trend of co-resistance in C. coli is suggested by the linear analysis, but it has shown an increase in 2024 compared to 2023, mirroring the C. coli pattern of tetracycline resistance on its own.
6.7.4. Human disease isolates from Wales
Unfortunately, only a few Campylobacter isolates could be recovered from the Welsh isolate collection from 2012 and 2013. The isolates were stored on beads and had been moved between institutions on two occasions which may have affected their long-term viability. For the most part, the clonal complexes most common amongst Oxfordshire disease were the most common amongst the Welsh isolates also. These were namely ST-21CC, ST-828CC, ST-48CC, ST-464CC and ST-45CC, though ST-48CC was not identified amongst the eight Welsh isolates from 2013. Two C. coli isolates were isolated each year, the remainder were C. jejuni.
Fluoroquinolone resistance was predicted for 11/33 (33.3%) of the C. jejuni isolates from 2012 and 3/6 (50%) of the C. jejuni isolates from 2013, for which the full coding sequence of gyrA was recovered. Tetracycline resistance was predicted for 11/33 (33.3%) of the C. jejuni isolates from 2012 and 3/6 (50%) for the C. jejuni isolates from 2013. Resistance to both antimicrobial classes was predicted for 8/33 (24.2%) of the C. jejuni isolates from 2012 and 2/6 (33.3%) of the isolates from 2013. One C. coli isolate from 2012 was fluoroquinolone resistant and the other was resistant to both fluoroquinolones and tetracycline. No fluoroquinolone or tetracycline resistance determinants were detected amongst the two C. coli isolates from 2013.
The T86I point mutation of the gyrA gene was the most common fluoroquinolone resistance determinant detected, with the D90N mutation detected in a single isolate (ST-607 complex) from 2012.
No macrolide or aminoglycoside resistance determinants were identified amongst the Welsh isolates.
6.8. Fluoroquinolone and tetracycline resistance amongst AgriFood isolates
Resistance determinants were more commonly detected for tetracycline than fluoroquinolones amongst the AgriFood and environmental isolates (Figure 21). The highest level of 57.2% tetracycline resistance was seen amongst isolates from the free-range broiler breeder flock sampled in 2004/5 (note, this was before the antibiotic stewardship scheme by the British Poultry Council was introduced). Tetracycline resistance was above 33% amongst the other commercial chicken sources tested, followed by backyard chickens with 28.1% resistance. Tetracycline resistance determinants were detected amongst 21.7% of Campylobacter isolates from ducks. Lower levels of resistance were seen amongst the starlings (17.4%) and geese (1.0%). It should be noted that the number of isolates from starlings in this study was relatively few, with the resistant isolates from starlings belonging to ST-21CC, ST-443CC, ST-828CC and ST-1150CC. These complexes are more commonly associated with livestock and are over-represented amongst the starlings in this study compared to the natural population as isolates were selected for testing to represent Campylobacter lineage diversity rather than prevalence. The ST-177CC and ST-682CC that make up the majority C. jejuni population from starlings were not associated with AMR determinants. Tetracycline resistance was not detected amongst the Campylobacter isolates from cattle and sheep.
Fluoroquinolone resistance was highest amongst the poultry sources and most commonly detected amongst isolates from the broiler chickens (32.0%), followed by the housed broiler breeder flock (22%), farmed ducks (16.9%), free-range broiler breeder flock (15.4%) and backyard chickens (11.5%). In contrast, isolates with resistance to fluoroquinolones was lower amongst cattle (2.1%), sheep (1.3%) and wild birds (starlings, 4.35% and geese 1%). Resistance is likely to be an over-estimate for starlings, as for the tetracycline.
Resistance to both fluoroquinolones and tetracycline was seen only amongst poultry, with levels highest amongst the broiler chickens (30%), followed by housed broiler breeders (18.8%), farmed ducks (16.9%) and free-range broiler breeders (3.2%). Co-resistance to the two antibiotics was associated with the C. coli ST-828CC (46/92, 50%) and C. jejuni ST-464CC (12/92, 13%), ST-353CC (3/92, 3.1%), ST-573 (2/92, 2.2%) and ST-354CC (2/92, 2.2%) amongst chickens, and ST-574CC (14/92, 15.2%) amongst ducks.
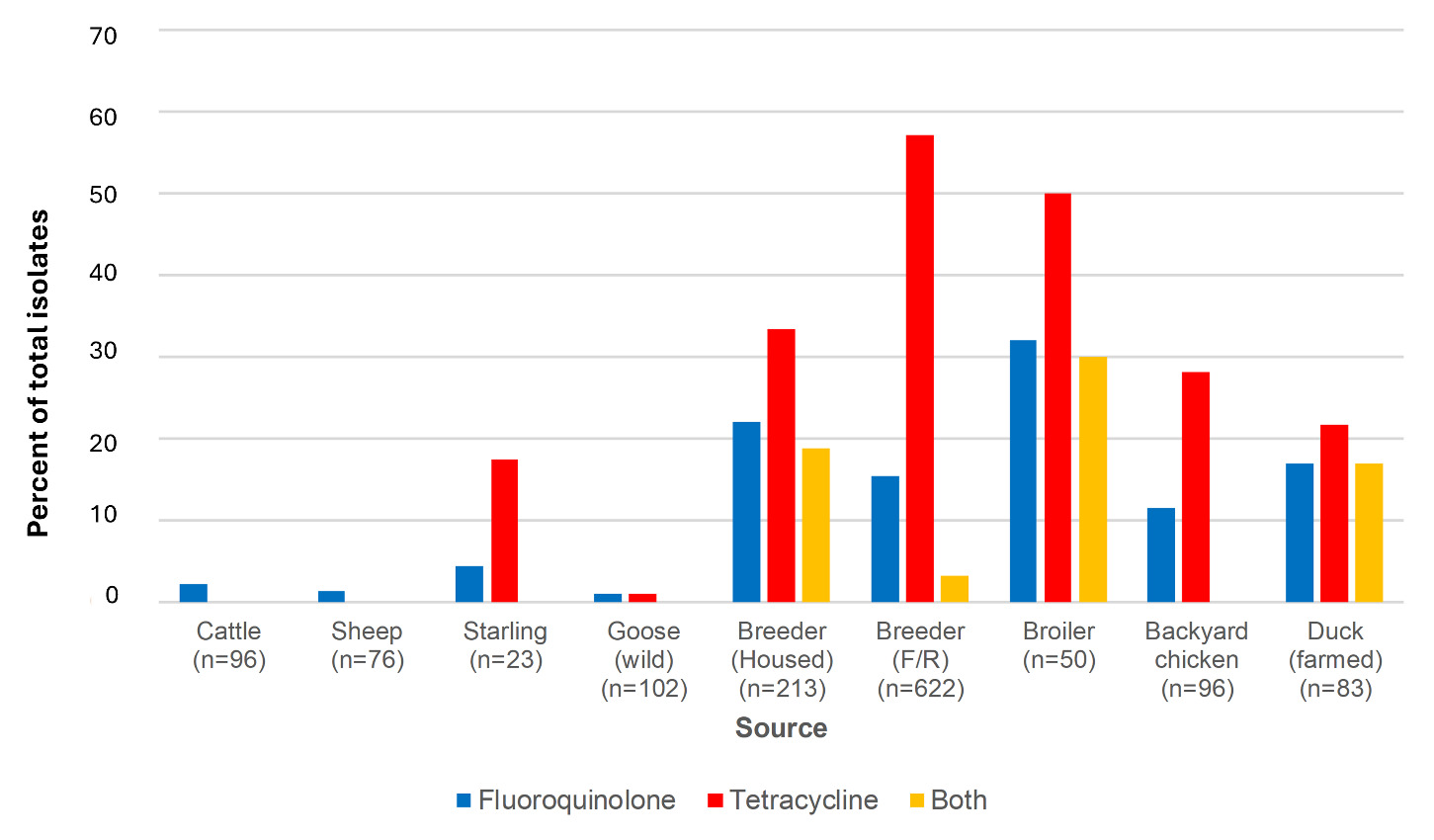
6.9. Campylobacter lineages associated with antimicrobial resistance
6.9.1. Association of fluoroquinolone and tetracycline resistance determinants with clonal complex; human disease isolates
The following clonal complexes showed significant association with fluoroquinolone resistance (p <0.01); ST-21CC, ST-52CC, ST-353CC, ST-354CC and ST-464CC, together with C. jejuni isolates that were unassigned to a clonal complex (Figure 22, Table 25, Appendix A). Highest resistance was seen amongst ST-464CC (98%), ST-354CC (95.7%) and ST-353CC (91.3%). These three clonal complexes are known to be chicken-associated, cross-checked by exploration of the global collection of Campylobacter isolates on the PubMLST database and the aiSource (Arning et al., 2021) modelling algorithm which gives the following estimates of chicken source for the Oxfordshire human disease isolates: ST-464CC (65.7%), ST-354CC 86.4% and ST-353CC (91.3%).
The following clonal complexes showed significant association with fluoroquinolone sensitivity (p<0.01); ST-22CC, ST-42CC, ST-45CC, ST-48CC, ST-61CC, ST-206CC, ST-257CC, ST-283CC, ST-443CC, ST-508CC, ST-658CC and ST-828CC. ST-257CC is known to be chicken associated, with aiSource estimating that 98.2% of Oxfordshire human disease ST-257CC isolates are derived from this source.

The following clonal complexes showed significant association with tetracycline resistance (p < 0.01); ST-354CC (97.9%), ST-464CC (97.5%) and ST-353CC (88.4%) (Figure 23, Table 26, Appendix A). The following clonal complexes showed significant association with tetracycline sensitivity (p <0.01); ST-22CC, ST-42CC, ST-45CC, ST-49CC, ST-61CC, ST-283CC, ST-403CC, ST-508CC and ST-828CC.

6.9.2. Association of fluoroquinolone and tetracycline resistance determinants with clonal complex; AgriFood and environmental isolates
Fluoroquinolone resistance was significantly associated with ST-21CC, ST-353CC, ST-464CC, ST-574CC (Figure 24, Table 27 Appendix A). Tetracycline resistance was significantly associated with ST-443CC, ST-464CC, ST-573CC, ST-574CC, ST-607CC, ST-661CC, ST-692CC and the C. coli ST-1150CC (Figure 25, Table 28 Appendix A).


6.9.3. Fine typing using Life Identification Number (LIN) barcoding demonstrates stability of fluoroquinolone resistant lineages over time
A couple of examples of the most prevalent C. jejuni STs associated with fluoroquinolone resistance amongst the Oxfordshire human disease isolates from this study are shown below (Figure 26). Identical sub-lineages, matching at all of the 1,142 cgMLST v2 loci are highlighted with distinct colours in the bar chart, and identified by specific LIN bar code. Note, the majority of isolates shown were also tetracycline resistant, but there were exceptions within each of the identical sub-lineages.
The longevity and global spread of these particular sublineages were further explored amongst the global collection of more than 99,000 Campylobacter isolates with genome data on the PubMLST database. Most sub-lineages within the threshold of 1 different cgMLST v2 locus were confined to the isolates in this study. ST-6175 (ST-21CC), LIN code 0_0_0_0_1_6_0_0_0_0_2_0_1_0_0_0_0_8, was an exception however. For this LIN code, isolates with just one cgMLST v2 locus different to those in this study were found amongst isolates from years 2016-2024, predominantly from the UK, but also from The Netherlands, France, and Denmark. Isolates were from human disease or source unknown, together with one from chicken and one from cattle. The global spread of other AMR lineages will be investigated further.
6.10. Genome Association Study
In order to test whether Campylobacter isolates grouping into the chicken-associated clonal complexes with high level AMR pose an increased risk to human health, we screened those with high prevalence resistance (ST-353CC, ST-354CC and ST-464CC) for presence of genes associated with aerotolerance, biofilm formation and virulence, and compared them with ST-257CC isolates with lower AMR prevalence (Gharbi et al., 2023; Pokhrel et al., 2022). Two of the genes tested, htrA and sodB are associated with both aerotolerance and biofilm formation, and indeed there may well be further genetic overlap between the two phenotypes.
In addition, we ran a pilot study to compare aerotolerance phenotype amongst lineages with high and low AMR.
6.10.1. Aerotolerance, genomic screen
We screened isolates for the following genes associated with aerotolerance; Alkyl hydroperoxide reductase-Antioxidant (ahpC), Catalase (katA), Iron co-factored superoxide dismutase (sodB), Ferredoxin A-cyclophilin gene (fdxA), High temperature requirement-A protease (htrA), Thiol peroxidase (tpx), Truncated hemoglobin (ctb) and MarR family transcriptional regulator Cj1556 (Pokhrel et al., 2022).
Alleles were generally associated with clonal complex for each of the genes tested. Two genes, fdxA (Figure 27) and htrA (Figure 28) had unique alleles found in ST-257CC isolates, compared to isolates from ST-353CC, ST-354CC and ST-464CC. When comparing the fdxA and htrA alleles of interest across the rest of the isolates in this study, none that were isolated with frequency >10 was exclusively associated with either AMR sensitive or resistant isolates.
_alleles_amongst_chicken_associated_clon.svg)
_alleles_amongst_chicken_associate.svg)
6.10.2. Aerotolerance, phenotyping pilot study
6.10.2.1. Introduction
A subset of 130 Campylobacter isolates was chosen to represent different resistance profiles across multiple lineages and tested for aerotolerance and survival in an aerobic atmosphere. The aim was to investigate the level of risk they may pose through stages of meat processing, environmental exposure and to the human consumer in their kitchen. A simple experimental procedure was used to test for variation and evaluate the need for further investigation using more sophisticated methods.
6.10.2.2. Method.
The Campylobacter isolates were recovered from frozen stocks by inoculating them onto Columbia Blood Agar, which was incubated at 41°C +/- 1°C in a microaerobic atmosphere for 24-48 hours. A single colony from each plate was then sub-cultured onto a fresh Columbia Blood Agar plate and incubated microaerobically at 41°C +/- 1°C for 24 hours. The agar plates were then left in aerobic conditions at room temperature (20-22°C) on the laboratory bench to mimic Campylobacter contaminated food being left on a kitchen surface. The agar plates were left for up to 120 hours (5 days), though none of the isolates tested in this study were viable beyond 96 hours (4 days). Viability (survival in aerobic conditions) for each isolate was determined by sub-culturing a single colony onto fresh Columbia Agar plates at 24-hour intervals, incubated at 41°C +/- 1°C in a microaerobic atmosphere for up to 48 hours and checking for presence/absence of growth.
Results were shown to be reproducible using the Fleiss Kappa test (p <0.05) for five Campylobacter isolates, for which the assay was repeated three times. In order to capture the diversity of lineages, and also for reasons of cost and time efficiency, we therefore chose to test multiple isolates within and between lineages, rather than repeat the phenotyping for all individual isolates multiple times.
6.10.2.3. Results and discussion
Detailed results and isolate information can be seen in Table 30 in Appendix A. Despite Campylobacter being a difficult organism to culture once outside its intestinal niche, there was variation in survival times for isolates, from less than 24 hours up to 96 hours exposure in an aerobic atmosphere (Figure 29). Isolates from the environment associated ST-1034CC and ST-45CC had shortest survival times of <24 hours, whilst ST-354CC and the C. coli ST-828CC isolates had longest survival times of 96 hours. (Note, ST-45CC isolates were annotated as environmental isolates in this experiment reflecting their frequent isolation from wild birds and environmental sources, though they are also recognised to be multi-host associated). Just over half of the isolates (73/130, 56.2%) could be recovered after 24 hours aerobic exposure.
Chicken associated isolates, which represent the major source of human infection were able to survive in aerobic conditions for up to 96 hours. In addition, AMR isolates were able to survive for up to 96 hours aerobically, including one C. coli isolate resistant to three classes of antimicrobials that was recoverable after 96 hours.
_clonal_complex__(.png)
Comparison of phenotype and genotype found some gene alleles may predict aerotolerance, shown in dark blue in the heat map in Figure 30. A Multiple Correspondance Analysis found fdxA allele 3, ahpc allele 3, ctb allele 6, tpx allele 16, and sodB allele 11 to be the top contributing alleles to the first principal component (PC1), but they only explained 4% of the variation. Fisher’s Exact test results showed a significant relationship above 48h for all 5 alleles (p<0.05).
The results indicate that C. coli pose the biggest risk for survival outside of niche host-associated conditions, but multi-host (ST-21CC) and chicken associated C. jejuni, as well as AMR lineages, were also able to survive aerobically for 24 hours or more. Environmental lineages were the most sensitive to being left on the bench, with most unable to be recovered after 24 hours. It could be hypothesised that Campylobacter lineages better able to persist outside the host source, and/or on kitchen surfaces, would lead to greater human exposure and prevalence amongst human disease.
Whilst care was taken to make the test conditions as consistent as possible, the results are qualitative rather than quantitative and a subset of predominantly resistant isolates were tested. In addition, whilst the Campylobacter cultures contained many millions of bacterial cells, contamination levels in the natural environment are likely to be considerably lower. Further work is needed to determine mechanisms for aerotolerance, in order to understand what is likely to be a complex genotype.
6.10.3. Biofilm formation
We screened isolates for the following genes associated with biofilm formation; Glycosylated structural flagellins-A (flaA, short variable region), S-ribosylhomocysteianse (luxS), Campylobacter adhesion to fibronectin (cadF) Chaperone DnaJ (dnaJ), High temperature requirement A (htrA), Superoxide dismutase (sodB), Alkyl hydroperoxide reductase (ahpC), a homolog of cluster 3 binding protein (peb4), Thioredoxin A (trxA), Thioredoxin B (trxB), Branched chain amino transferases for leucine, isoleucine, and valine (ilvE) and NuoC- subunit of complex I (ubiquinone oxidoreductase) (nuoC) (Pokhrel et al., 2022). Only the cadF gene had alleles that were unique to ST-257CC isolates, in comparison to isolates from ST-353CC, ST-354CC and ST-464CC (Figure 31).
_alleles_amongst_chicken_.svg)
6.10.4. Virulence determinants
Cytolethal Distending Toxin (CDT) is an important virulence factor in C. jejuni, which causes cell death through the active DNAse activity of the CdtB subunit, and is associated with local inflammation (Lai et al., 2016). We compared genetic elements encoding three protein subunits that form the CDT amongst the ST-353CC, ST-354CC, ST-464CC and ST-257CC isolates in this study.
We also looked for presence of the wlaN and cgtB genes that are associated with Guillain Barré Syndrome (GBS), a polyneuropathic disorder that can follow Campylobacter infection (Guirado et al., 2020; Linton et al., 2000). The genes encode a β-1,3-galactosyltransferase enzyme which produces sialyated lipopolysaccharide that mimics human GM1 ganglioside present in peripheral nerves. Antibodies directed against the Campylobacter sialyated lipopolysaccharide may then cross-react with human GM1 ganglioside, resulting in GBS.
A total of 26 cdtA-cdtB-cdtC profiles were identified amongst the ST-257CC, ST-353CC, ST-354CC and ST-464CC (Figure 32). With the exception of cdtA-cdtB-cdtC 6-9-1, identified amongst the ST-353CC and ST-464CC, the profiles did not overlap. Each clonal complex was associated with one predominant cdt toxin profile, but not exclusively. Similarly, with the exception of CDT profile 3-9-3 associated with ST-257CC, the toxin profiles were not unique to the clonal complexes within the wider dataset, and were occasionally linked with isolates grouping into different clonal complexes. Different toxin profiles within a clonal complex were most commonly linked with ST, and were consistent across sources where more than one existed.
No wlaN or cgtB genes were identified amongst any of the ST-257CC, ST-353CC, ST-354CC or ST-464CC isolates.
In the wider dataset, 6 wlaN gene alleles were identified amongst 51 C. jejuni isolates (1.3% of the dataset), of which 32 were predicted to have a functional protein, recognised by 8 G’s within a homopolymeric G-tract (Guirado et al., 2020; Linton et al., 2000). Other variants found in this study had 8, 10 and 11 G’s within the homopolymeric tract. Isolates with predicted functional wlaN protein were most commonly isolated from human disease (21/32, 65.6%) and chicken (9/32, 28.1%), including housed broiler breeders, backyard chickens and a broiler chicken, with just one isolate each from ducks and cattle. The isolates grouped into ST-21CC (21/32, 65.6%), ST-573CC (6/32, 18.8%) and ST-206CC (5/32, 15.6%).
Additionally, 7 cgtB alleles were identified amongst 132 C. jejuni isolates (3.3%) of the wider dataset. They were most commonly associated with ST-42CC (n=57), ST-206CC (n=36), ST-22CC (n=18), ST-52CC (n=10), ST-508 (n=4), ST-21 (n=4), ST-460 (n=1), with 2 isolates unassigned. Most isolates were from human disease (n=88), followed by sheep (n=19), housed broiler and broiler breeder chicken (n=18), cattle (n=6) and geese (n=1). All isolates from the animal sources were from ST-42CC, with the exception of one ST-22CC isolate from a backyard chicken.
Highest risk of GBS associated wlaN or cgtB genes were identified amongst isolates from ST-42CC (57/121, 47.1%), followed by ST-206CC (41/135,30.4%), ST-22CC (18/67, 26.9%), ST-460CC (1/4, 25%), ST-508CC (4/19, 21.1%), ST-573CC (6/38, 15.8%), ST-52CC (10/72, 13.9%) and ST-21CC (25/793 ( 3.2%). No C. jejuni isolates in the study contained both wlaN and cgtB genes. No wlaN or cgtB genes were identified amongst C. coli isolates in the study.
Of the 164 isolates from the study that were found to have GBS associated wlaN and cgtB genes, 34 (20.7%) were resistant to fluoroquinolones and 21 (12.8%) were additionally resistant to tetracycline. None were found to have resistance determinants for macrolide or aminoglycosides.
Previous studies have found GBS causing C. jejuni to be associated with ST-22CC and ST-403CC (Hayat et al., 2023; Islam et al., 2009; Nielsen et al., 2010). ST-206CC (and in addition ST-922CC, ST-22CC and ST-48CC) has been linked with greater risk of post Campylobacter infection Irritable Bowel Sydrome (Peters et al., 2021).

7. Discussion
7.1. Study limitations
It should be noted that the in some cases (starlings or deer for example) the number of isolates is relatively small and do not represent a complete population study. The study focused on acquiring whole genome sequencing data from sources of Campylobacter that have been under sampled to date. This includes live animals on farms as the origin of infection, rather than later steps of food production which may be more conveniently sampled.
Given the challenges to obtaining representative national coverage, a pragmatic approach was taken to use existing and ongoing isolate collections. Some are historically valuable and capture the era pre concerted antimicrobial stewardship that can be compared with contemporary collections.
Whilst efforts have been made to capture as wide diversity as possible from each of the sources, it is not practical to sample very large numbers of farms, and instead, multiple animals have been sampled from the same flock, herd or farm. As a result, some flock/herd or farm-associated STs will be over represented in the dataset.
It should be noted that isolates from Oxfordshire human disease begin in April 2019, and that isolates from July-September 2021 contributed to a study by the UKHSA (Swift et al., 2025) and are absent from this study.
The AMR phenotyping results generally show good agreement with genotypic prediction, but there will always be challenges regarding quality of WGS data and completeness of sequencing assemblies. Results using PubMLST are shown for full coding gene sequence from assembled sequencing data. Genetic determinant nomenclature for the AMR varies in the literature and across different AMR finder databases, with some determinants having more than one name. The AMR finder databases also vary in the AMR genetic variants they search for. Results from this study should be interpreted with the view that the AMR levels are conservative estimates, predicted from genomic data, but are in line with findings from our previous study (Oxford, 2019).
7.2. Source attribution.
Each of the AgriFood and environmental sources tested represented a potential source of human infection, having at least one isolate grouping into the C. jejuni ST-21CC, ST-45CC and C. coli ST-828CC that are amongst the most common human disease-causing clades. These clonal complexes also had the broadest distribution across 11 (ST-828CC to 12 (ST-45CC) of the different sources tested, demonstrating their success at colonising different hosts. As expected, other isolates grouped into host-associated clonal complexes for the most part. For example, ST-61CC was identified amongst cattle and sheep isolates, and ST-353CC and ST-354CC were identified amongst the chicken isolates, as seen in other studies. Of the Scottish sources tested, ST-61CC was identified from deer for which little information exists, and ST-1034CC was common amongst wild geese grazing on farmland, matching findings from Oxfordshire geese sampled previously (Colles et al., 2008).
Population studies of live chickens from breeder flocks, extensively reared broiler (free-range meat chicken) flocks and backyard chickens are rare, and were included in the study to give valuable nuanced information regarding the epidemiology of Campylobacter in the most predominant source of human infection. Extensively reared chickens in particular cannot by definition be kept under as strict biosecurity measures as housed birds and will be exposed to environmental sources of infection, whilst breeder flocks are subject to the strictest controls possible.
Overall, a number of chicken-associated clonal complexes were found in each of the chicken sources despite their differing management. This suggests that host association is in part driven by Campylobacter lineage, with a number of clonal complexes being maintained in seemingly separate types of poultry. With such restricted access, it is difficult to comprehend how Campylobacter from breeding flocks would reach the human consumer however, and further high resolution typing using LIN code analysis is needed to confirm whether or not this is the case. There is widely held consensus that vertical transfer of Campylobacter from parent breeding flocks to offspring (broiler/meat chickens) does not occur since Campylobacter cannot be isolated from eggs (Callicott et al., 2006; Rossi et al., 2012; Shanker et al., 1986), but it is difficult to completely rule out vertical or horizontal transfer between flocks by other routes such as fomites on equipment (Cox et al., 2012; Sahin et al., 2017). Such events may still be rare, and when dealing with millions of animals produced each week (DEFRA, 2025), diverse Campylobacter lineages and good biosecurity practises, precise evidence is difficult to obtain.
As might be anticipated, clonal complexes more commonly associated with environmental sources were occasionally isolated from the extensively reared chicken flocks but not from intensively reared broiler chicken flocks, indicating that well executed biosecurity measures for housed birds are effective in excluding infection from these sources. These include ST-682CC, ST-1034CC and ST-1275CC.
Isolates grouping into ST-661CC were common amongst the free-range broiler breeder flock, but were not found amongst human disease. The lineage is thought to poorly survive meat processing steps, but the results may also reflect the small market share of free-range poultry meat. It is of note that isolates from this particular complex were associated with tetracycline resistance, despite less intensive rearing conditions. The highly resistant chicken associated ST-464CC was not isolated from the free-range broiler breeder flock, or the backyard chicken flocks, though these isolate collections are older and not contemporary.
We identified horses to be an under-sampled potential source of infection for human disease, with no isolates included on the PubMLST database at the time of this study. We have increased the number to two isolates from horses that were sick, but despite our best efforts to isolate Campylobacter from healthy horses in order to genotype them, it was not possible within the time frame available. It was concluded that in comparison to other livestock where Campylobacter can be more easily be isolated using standard methods, healthy horses represent an unlikely source of human infection, if at all. It is also rare to isolate Campylobacter from sick horses; however, infected horses could present an infection risk to humans and need to be handled hygienically. The decision was taken that it would take considerable effort and cost to continue sampling this host source, which could be better directed to better understanding major routes of transmission where more significant impact could be made.
Our previous study in 2015-18 gave the best estimate of 70% of human infection from Oxfordshire and the North-East of England to originate from poultry sources using a sentinel approach and a combination of Structure (Pritchard et al., 2000) and iSource (Wilson et al., 2008) modelling algorithms, corrected for bias identified in self-attribution. Since this period, there has been success in reducing the number of most highly contaminated chicken carcasses with >1000 colony forming units (CFU) per gram at slaughter in the UK from 27% in 2008 to less than 10% in 2016 amongst major retailers (Jorgensen et al., 2019). The period of this study also spans the COVID-19 pandemic associated lockdowns and significant changes in human behaviour with regards to travel and eating out in 2020 and 2021 (see also section 8.6).
We used the aiSource (Arning et al., 2021) algorithm in this study, which has improved accuracy over iSource (Wilson et al., 2008) used in our previous study, due to a machine learning algorithm and ability to use cgMLST data over 7-locus MLST (Arning et al., 2021). We have further refined the published aiSource algorithm to use the more broadly applicable cgMLST v2 typing scheme, an updated training data set that does not seek to overdiscriminate between ruminant or rare sources of infection, and to model source attribution of C. jejuni and C. coli separately for greater accuracy.
Despite success in reducing the prevalence of the most highly contaminated chicken carcases at slaughter, our latest model predictions estimate that more than 80% of UK Campylobacter infection originates from poultry, followed by ruminant sources which account for most of the rest. The results indicate more work and investment is needed if we are to reduce the levels of campylobacteriosis, which have been higher than ever in 2024 (Agency, 2025). With chickens predominantly colonised with chicken associated lineages, it is still uncertain how they spread within the poultry industry. Until this is known, it is difficult to recommend intervention strategies beyond biosecurity measures and careful management of poultry flocks that would be broadly applicable to all pathogens ((JEMRA), 2023).
7.3. Antimicrobial resistance
7.3.1. Oxfordshire human disease C. jejuni isolates
For C. jejuni, fluoroquinolone resistance has increased from 45% in our last study in 2015-2018 to 55.2% in 2024 and has almost doubled since the start of our Oxfordshire human surveillance study in 2003 (28.2% fluoroquinolone resistance). However, the prevalence is lower than some reports from other regions of the world (Bort et al., 2022; Giacomelli et al., 2014; Jeong et al., 2025; Maesaar et al., 2016; Qin et al., 2023), and it has remained around 55% for the past three years, demonstrating the importance of antimicrobial stewardship in UK agriculture. Continued vigilance is recommended since there appears to have been stepwise changes in fluoroquinolone resistance over the years and further increases in resistance remains unpredictable.
The results for tetracycline resistance were more consistent amongst C. jejuni, with 30-50% of isolates carrying the tetO gene. Further research is needed to investigate if the gene is carried on a plasmid or chromosome, but results suggest that there is either low fitness cost to the bacterium, or continued selective advantage for tetracycline resistant lineages to persist, whether or not it is directly related to tetracycline resistance.
A high proportion of C. jejuni isolates were resistant to both fluoroquinolone and tetracycline, with the highest level of 43% recorded in 2024, and trends mirroring that of fluoroquinolone resistance over time. Particularly concerning is the very high-level resistance to fluoroquinolone (88-99.7%) and tetracycline (85.7-100%) amongst isolates grouping into the ST-353CC, ST-354CC and ST-464CC that are known to be chicken-associated. ST-353CC isolates were the most likely to be resistant to three classes of antimicrobials tested in this study. ST-354CC isolates were amongst the most aerotolerant in the phenotyping pilot study, indicating that they could pose additional risk of causing infection and complications with treatment if they are able to linger on surfaces, although they were the least prevalent of these three clonal complexes in Oxfordshire human disease being 14th most common overall.
It is difficult to predict why these clonal complexes should be so highly resistant, given other chicken associated Campylobacter lineages, such as ST-257CC, have much lower resistance. Comparison with the global collection of isolates on the PubMLST database, shows the three highly resistant lineages have been isolated internationally and sometimes across several continents. There are many factors that could affect the survival of these lineages and co-selection for AMR or otherwise, from colonising live birds on farms, through to the multiple stages of meat processing and food preparation, and further investigation is warranted. Fluoroquinolone resistance was lower (61.8%) but also significantly associated with ST-21CC isolates, which is also of concern due to the more generalist ability of these isolates to infect multiple hosts and potentially spread resistance further.
Aminoglycoside resistance ranged from 4-20 isolates per year (1.2-4.5%) with an overall prevalence of 3.1% (47/2585) in this study, showing an increase from 1% resistance in 2015/18 (Oxford, 2019). Macrolide resistance was not recorded amongst C. jejuni isolates in the 2015/18 study, but was predicted for 1-4 (0.2-0.8%) isolates a year in 2019-2024 and a total of 13/2585 (0.5%) of C. jejuni isolates in this study. The emerging ermB gene (Bolinger & Kathariou, 2017) encoding high resistance for macrolides was not detected amongst isolates in this study, but a subset of isolates with a 23S rRNA point mutation tested phenotypically were nevertheless demonstrating the highest levels of erythromycin resistance. Genetic determinants of a resistance enhancing (RE) cmeABC efflux pump that increases ciprofloxacin (fluoroquinolone) resistance 9-fold and erythromycin (macrolide) resistance 4-fold (Schiaffino et al., 2024; Yao et al., 2016) were detected amongst 8.6-10.2% of C. jejuni isolates each year. Further work is needed to fully explore links of these genetic determinants with AMR phenotype.
The results demonstrate that despite improved antimicrobial stewardship in both livestock and clinical settings, fluoroquinolone and tetracycline resistance continues to be a significant issue. Resistance to macrolides and aminoglycosides remains low, but has increased slightly since our last study nevertheless, emphasising the need for continued vigilance with antimicrobial stewardship. Resistance to three of the antimicrobial classes (fluoroquinolones, tetracycline and macrolides) tested in this study was detected in 13/2585 (0.5%) C. jejuni isolates, which were most commonly from ST-353CC. The impact of foreign travel and global trends in Campylobacter AMR are outside the scope of this study, but higher levels of resistance in other regions of the world also demonstrate the need for continued careful control of antimicrobial use, in order to preserve their efficacy for treating severe infections.
7.3.2. Oxfordshire human disease C. coli isolates
The numbers of C. coli recovered each year were approximately 10% that of C. jejuni, and matches that of our previous study and others (Graham et al., 2024; Oxford, 2019). In this study, we found fluoroquinolone and tetracycline resistance to be higher in C. coli human disease isolates than C. jejuni in 2003, but the trend is now reversed, with highest resistance currently seen amongst C. jejuni isolates. Fluoroquinolone and tetracycline resistance levels have remained largely unchanged amongst C. coli isolates at approximately 30-45% resistance to fluoroquinolones and 20-40% resistance to tetracycline over the past two decades, though there is some fluctuation across individual years, perhaps due to the smaller number of isolates. Approximately half the fluoroquinolone resistant C. coli isolates were also resistant to tetracycline.
In contrast to fluoroquinolone and tetracycline resistance, predicted macrolide (0.04% C. jejuni, 4% C. coli) and aminoglycoside (1.9% C. jejuni, 12.0% C. coli) resistance was higher amongst human disease C. coli isolates than C. jejuni isolates. C. coli isolates were also the most likely to be resistant to three of the four antimicrobial classes tested in the study, though this accounted for just 12/2585, 0.5% of human disease isolates. Multi-resistance was most commonly to fluoroquinolones, tetracycline and macrolides, with one C. coli isolate resistant to fluoroquinolones, macrolides and aminoglycosides but not tetracycline. One C. coli isolate (1/300, 0.3%) was resistant to all four antimicrobial classes tested in this study.
Factors driving AMR in C. coli compared to C. jejuni are not understood.
7.3.3. Human disease isolates from Wales
It is unfortunate that the isolate collection from Wales had limited viability, and results need to be interpreted with caution given the small number. The clonal complex distribution was similar to that seen amongst Oxfordshire disease, indicating that the source of human infection is similar for both countries. Predicted resistance amongst the Welsh C. jejuni isolates in 2012 (33.3% fluoroquinolone, 33.3% tetracycline and 24.2% co-resistance) was broadly similar to the levels in Oxfordshire in 2012 (38.9% fluoroquinolone, 42.3% tetracycline and 27.2% co-resistance). Unfortunately, it is not possible to make any further conclusions based on the small number of isolates.
7.3.4. AgriFood, animal and environmental sources
In general, much lower rates of AMR were found amongst Campylobacter isolates from ruminant, wild bird and environmental sources compared to each of the poultry sources. As with human disease isolates, very high levels of resistance were seen amongst the ST-353CC, ST-354CC and ST-464CC isolated from poultry. The mechanisms of resistance matched those seen amongst the human disease isolates, with the Thr-86-Ile mutation and tet(O) genetic determinants being the most common. Resistance determinants were most commonly linked to ST and/or clonal complex across different host sources. For this reason, the prevalence of resistance amongst starling isolates is likely an over-estimate in this study where livestock associated STs were tested, which are outliers within the wider population sample. We did not detect macrolide resistance amongst AgriFood and environmental isolates. One C. jejuni isolate from cattle, and 63 C. coli isolates were predicted to be aminoglycoside resistant. The majority of aminoglycoside resistant C. coli isolates were from broiler breeder flocks (housed and free-range), with two isolates from sheep and one from a starling. They were restricted to five STs, many sampled more than once from the same flock. Twenty aminoglycoside resistant C. coli isolates were additionally resistant to fluoroquinolones and tetracycline. No AgriFood and environmental isolates were resistant to all classes of antimicrobials tested in the study.
Taken together, the results from this study indicate that efforts to reduce the impact of zoonotic spread of AMR Campylobacter should concentrate on poultry sources of infection, though of course continued vigilance for other sources is still recommended. It is also recommended that efforts are continued to monitor spillover of AMR from intensive agriculture to the environment.
7.4. Genome association study
The prevalence of ST-353CC, ST-354CC and ST-464CC with elevated levels of AMR is concerning, and we compared genetic determinants associated with aerotolerance, biofilm formation and virulence with isolates from ST-257CC with lower AMR prevalence. Each of the clonal complexes tested are associated with chicken sources and genetic differences may provide clues as to their survival through meat processing, or ability to cause disease. It was generally found that alleles for each of the genes tested were associated with different lineages based upon 7-locus MLST, but were not exclusively associated with AMR resistance or sensitivity when comparing them across the wider dataset. The Ferredoxin A-cyclophilin gene (fdxA) and High temperature requirement-A protease (htrA) genes associated with aerotolerance and the Campylobacter adhesion to fibronectin (cadF) genes showed greatest distinction between the clonal complexes tested with high versus low AMR. The aerotolerance phenotyping pilot study indicated that chicken associated lineages, together with C. coli, were able to retain viability over longer periods compared with environmental lineages. Two ST-354CC and eight ST-828CC isolates were able to survive for up to 96 hours on agar plates held at room temperature in aerobic conditions. Similarly, AMR isolates were able to survive up to 96 in the aerobic test conditions and further research exploring how these findings relate to risk in human infection would be valuable. All isolates within these clonal complexes contained genes encoding the cytolethal distending toxin (CDT), indicating they have potential to cause inflammatory enterocolitis.
More broadly amongst the wider dataset, the cdtA-cdtB-cdtC toxin profile was linked with ST and clonal complex across different host sources, although not exclusively. It was noted that CDT genetic determinants were commonly truncated or contained numerous indels for isolates from wild birds, environmental sources and further research is required to investigate this further.
Campylobacter infection is the most common infectious precursor of Guillain Barré Syndrome, a polyneuropathic disorder resulting from molecular mimicry of the human GM1 ganglioside in peripheral nerves (Guirado et al., 2020; Linton et al., 2000). Previous studies have found an association with Guillain Barré Syndrome (GBS) and isolates that contain wlaN or cgtB genes, most commonly from ST-22CC but also from ST-403CC (Islam et al., 2009; Nielsen et al., 2010). We detected the GBS associated genes amongst 164/4010, 4.1% of isolates from this study, with the wlaN gene most commonly found in chicken isolates, and the cgtB gene in isolates from ruminants. Of these isolates, 20.7% were predicted to be fluoroquinolone resistant and 12.8% tetracycline resistant, raising concerns over potential complications for treatment. We further identified ST-42CC from ruminants to be a particular risk, along with isolates from a handful of clonal complexes additional to the previously recognised ST-22CC and ST-403CC. Whilst the isolates may have potential to cause GBS based upon their genetic elements, the relatively low incidence of the syndrome following 1/1000 Campylobacter infections indicates that this does not always happen and there are likely to be a number of factors involved, including the human host immune reaction.
Genome association results presented here are a preliminary screen and more work is needed to elucidate the clonal complex interactions between phenotype and genotype in order that virulence and risk to human health can be better predicted. Genome Wide Association Studies (GWAS) may additionally be used as a tool to identify other genes of influence that can be tested in phenotyping studies.
7.5. Effect of COVID-19 lockdowns on human disease
We have previously established that results from the ongoing Oxfordshire human disease study mirrors that of a national scale (Oxford, 2019). There has been variability in reported incidence of campylobacteriosis across many countries during the COVID-19 pandemic (Liu et al., 2022). The lower numbers of PCR positive samples detected by the Oxfordshire clinical laboratory in April and September 2020 likely reflects reduced healthcare seeking behaviour and availability of medical appointments during the COVID-19 lockdowns, whilst the overall number of infections detected in 2020 remained high, in line with the other years covered by this study (Ondrikova et al., 2021). The overall distribution of clonal complexes was unchanged in 2020 compared to other years in the study and there was no measurable impact of restrictions on eating out or foreign travel during this time. The results of this study are consistent with the major source of campylobacteriosis in the UK being associated with chicken, though precise routes of transmission remain to be identified. For example, the comparative risks of home vs commercially prepared food, risk from under cooked meat vs cross-contamination in the kitchen, imported vs UK produced meat and the impact of different supply chains.
There were five isolates (5/361, 1.4%) identified in 2020, that unusually belonged to clonal complexes associated with wild birds and environmental sources (ST-1034CC, ST-1275CC and ST-1332CC) and could have resulted from outdoors activity. These clonal complexes had similar prevalence in 2022 (7/448, 1.7% of isolates) and 2024 (2/511, 0.4% of isolates), however, but were not seen in 2019, 2021 or 2023. Two isolates with usual fluoroquinolone resistance determinants, Thr86Ala and Thr86Val, one grouping into the environmentally and generalist associated ST-45CC and the other being unassigned, were identified in 2020. One further example of each was also detected in 2022 and 2024 amongst a C. coli and ST-52CC isolate. These isolates could potentially reflect interaction with rarer sources of environmental infection. Overall, results indicate that there was no greater association with environmental sources of infection during COVID-19 lockdowns than the rare events that occur stochastically in other years.
There was a significant increase of Campylobacter PCR-positive cases seen in June 2021, prompting national surveillance of Campylobacter infection by the UKHSA. The overall clonal complex distribution appeared unchanged relative to other years, but the significant increase in the number of cases could reflect the relaxation of lockdown rules, combined with seasonal summer peak of infection. The ST’s most commonly isolated during June 2021 were ST-45 (ST-45CC) (12/89, 13.5%), ST-6175 (ST-21CC) (8/89, 8.9%), ST-50 (ST-21CC) (n=5/89, 5.6%), ST-21 (ST-21CC) (n=5/89, 5.6%) and ST-48 (ST-21CC) (n=5/89, 5.6%). None were new to the study, having global distribution over the past decade amongst the extensive collection on the PubMLST database.
7.6. Utilising the PubMLST database
The PubMLST database provides globally recognised and definitive nomenclature for Campylobacter and more than 100 other microorganisms (Jolley et al., 2018). Data linking isolate metadata and curated genomic data are freely accessible. Public or private isolate databases allow users to store, analyse and explore data in a global context. Open-source analysis tools are integrated into the web-accessible interface, and BIGSdb (the software running PubMLST) functionality can be linked to other resources using Application Programming Interfaces (APIs) (Jolley et al., 2018).
As part of this study, we have continued to develop molecular typing functionality and detection of AMR resistance determinants (SNPs or amino acid variants) with user ease in mind. We have updated the cgMLST typing scheme together with newly developed stable lifetime identification number barcoding (LIN code) nomenclature that gives greater reliability and robustness for fine typing and identification of highly related lineages across C. jejuni and C. coli isolates from a wide variety of sources and locations. It is possible to assess presence/absence of genes or SNPs of interest across large isolate collections within seconds, and download sequencing data in a variety of formats required for downstream analysis. The data are fully curated, with the heavy lifting bioinformatics automated or already completed for many analyses.
7.7. Concluding remarks and recommendations
Cases of human campylobacteriosis in the UK are higher than ever and unlike some other pathogens, the incidence was unaffected by COVID-19 lockdowns. The results are consistent with the major source of UK Campylobacter infection being food consumed at home. The most accurate source attribution modelling to date estimates more than 80% of Campylobacter infection is associated with chicken, despite success in reducing prevalence of the highest contaminated birds at slaughter. Ruminants represent the second largest source, accounting for most of the rest of human disease cases.
Fluoroquinolone and tetracycline resistance remains high, and whilst relatively stable amongst C. jejuni over the past four years, further stepwise increases in resistance cannot be ruled out. Three chicken associated clonal complexes, ST-353CC, ST-354CC and ST-464CC have resistance to fluoroquinolones and tetracycline approaching 100%, for reasons unknown, whilst other prevalent chicken associated lineages show much reduced levels of AMR. At the current time, C. coli has slightly lower levels of resistance to fluoroquinolones and tetracycline, and this appears to be relatively unchanged over the past two decades, with exceptions in some years.
Preliminary results indicate that chicken associated lineages, as well as C. coli isolates, are more aerotolerant than environmental lineages, with some resistant isolates able to survive up to 96 hours on agar plates left on the bench. Further research is needed to confirm the results and assess the risk during food processing and to human disease.
Resistance to macrolides, aminoglycosides and multidrug resistance remains low, though levels have increased marginally since our last study in 2015-18. Resistance to macrolides was at the highest level tested phenotypically amongst isolates with a 23S rRNA point mutation, despite the ermB gene associated with high level macrolide resistance (Bolinger & Kathariou, 2017) not being detected. Genetic elements predictive of a resistance enhancing efflux pump seen in Campylobacter isolates from China and Peru were detected amongst 10% of isolates in this study, with implications still requiring to be validated.
Genetic determinants associated with Guillain Barré Syndrome were detected in 7% of isolates from the study, with the wlnA gene most common in poultry sources and the cgtB gene most common in ruminant sources. ST-42CC was highlighted as a particular potential problem, along with a subset of isolates from a handful of other clonal complexes. Information regarding GBS cases was not available for the human disease isolates in this study, but they are rare and expected to much lower than 7%. Further investigation of virulence factors is warranted given the increasing numbers of campylobacteriosis in recent years.
Results from this study indicate that antimicrobial stewardship appears to be working in the most part for macrolides and aminoglycosides, and may have slowed the rise in fluoroquinolone and tetracycline resistance compared to some countries, though levels remain high and three chicken associated clonal complexes (ST-353CC, ST-354CC and ST-464CC) are particularly challenging. Chicken continues to be the predominant source of human infection and a One-Health approach with interdisciplinary collaboration is needed to understand why a reduction in the most-highly contaminated chicken carcasses has not translated to a notable reduction in human disease cases. The nuances of imported meat and takeout versus home cooking are not yet quantified, but with Campylobacter being readily destroyed by cooking, interventions could also include social science based educational programmes. It remains, that greatest impact on the numbers of human infection will be achieved if transmission from chicken can be reduced.
The following recommendations are made to progress knowledge of Campylobacter and work towards reducing levels of human disease in the UK.
General recommendations
-
Campylobacter remains a major public health problem and the urgent need for research is underlined by it being an exemplar of a complex ‘One-Health’ pathogen.
-
Interdisciplinary, open, and collaborative approaches with stakeholders including industry are key to the understanding the complexity Campylobacter biology which is necessary to achieve disease control.
-
Field- and industry-based studies across the entire supply chain are essential to ensure the relevance of the science and for planning of future interventions.
-
Best information is achieved from representative and contemporaneous sampling of human, food and animals to aid understand transmission of Campylobacter.
-
High resolution molecular typing is necessary to ensure accurate epidemiology and source attribution.
Recommendations for research focus
Campylobacter based:
-
Fine typing (by cgMLST and LIN codes) should be used to determine how chicken-associated Campylobacter lineages spread across chicken production systems with the aim of designing more effective interventions.
-
The reason(s) why AMR Campylobacter lineages persist in the absence of antibiotic use on farms should be investigated.
-
Phenotyping of emerging antibiotic resistance mechanisms with complex and as-yet unconfirmed genotype, for example RE-cmeABE efflux pump and aminoglycoside resistance, is strongly recommended for accurate results.
Chicken focused:
-
More detail is needed regarding ‘chicken’ and poultry as a source of human infection. In particular, the extent to which infection is derived from consumption of under cooked meat versus cross-contamination during food preparation should be determined. It is currently unknown whether product type, imported meat, pre-prepared, take-out and home cooked foods give the same risk.
-
Better diagnostic tools for Campylobacter are needed for testing on-farm and in processing plants.
-
Measures to reduce Campylobacter contamination of chicken meat and live birds need to go beyond biosecurity and should be Campylobacter-focused for greatest impact. They also need to be practical, affordable and beneficial to both the consumer and industry if they are to be implemented. For example, improvements may be possible regarding chicken welfare and gut health (Colles et al., 2016, 2021; Rawson et al., 2020), optimised flock management (Rawson et al., 2021) across different settings, ‘smart farming’ technologies and promising but untested interventions such as Violet-Blue light (Walker et al., 2022) in meat processing.
Human focused:
-
Investigating which behaviour(s), population demographics, food and travel choices lead to the consumption of undercooked chicken meat or infection from cross-contamination.
-
Determining the use and effects of antimicrobials in the treatment of human infections and continuing to optimise best practise.
-
Investigating whether the ‘don’t wash chicken’ guidelines need to be updated in order to improve consumer hygiene and meat handling expertise.
-
Identifying whether particular groups are more susceptible to serious sequelae such as Guillain-Barré Syndrome, Reactive Arthritis, Irritable Bowel Syndrome or food intolerances following Campylobacter infection.
Campylobacter has been removed from the 2024 WHO Bacterial Pathogen Priority List which may give the false impression that Campylobacter related disease or AMR has fallen.
Campylobacter has been removed from the 2024 WHO Bacterial Pathogen Priority List which may give the false impression that Campylobacter related disease or AMR has fallen.
One Health: a unified approach recognising that the health of humans, animals (domestic and wild), plants the wider environment/ecosystems are closely linked and interdependent (WHO definition).